48uep6bbphidvals|153
48uep6bbphidcol4|ID
48uep6bbph|2000F98CTab_Articles|Fulltext
The discovery of Helicobacter pylori has completely changed our understanding of upper gastrointestinal diseases such as peptic ulcer and gastric cancer. Eradication of H. pylori has been recommended in treatment of peptic ulcer disease and primary gastric lymphoma. Epidemiological data suggest an overall decline in the incidence of peptic ulcer disease and an increasing incidence of gastro-oesophageal reflux disease and its complications such as Barrett’s oesophagus (BE) and oesophageal adenocarcinoma.(1,2) H. pylori infection is also decreasing worldwide due to better hygiene and increasing use of antibiotics.(3) Therefore, one may infer a possible negative relation between H. pylori infection and frequency and severity of gastro-oesophageal reflux disease based on these epidemiological data. Furthermore, it is important to address the role of host genetic factors in the pathogenesis of gastro-oesophageal reflux disease. Here we critically review the literature derived from PubMed searches using the key words gastro-oesophageal reflux disease, H. pylori, host genetic factors, Barrett’s oesophagus, and oesophageal adenocarcinoma. Based on these data, the relationship between gastro-oesophageal reflux disease, H. pylori and host genetic factors is reviewed, the possible mechanisms explaining their relationship is discussed and future directions for research outlined.
What may be the relationship between gastro-oesophageal reflux disease and H. pylori?
The pathogenesis of gastro-oesophageal reflux disease is multifactorial; various factors that may play important roles can be classified into host, environmental and dietary factors (Figure 1).(4,5) However, the exact role of H. pylori, host genetic factors and the interplay between these two in the pathogenesis of gastro-oesophageal reflux disease has still not been established.(6,7) Most studies suggest that the presence of H. pylori is protective against gastro-oesophageal reflux disease.(2,6,8,9,10,11) These studies have been reviewed below. However, a few studies suggested that H. pylori infection may exacerbate gastro-oesophageal reflux disease,(12,13,14) especially in patients with duodenal ulcer (DU),( 12,13,14) in whom gastritis is antral-predominant. Antral predominant gastritis increases gastric acid secretion. Also, H. pylori infection in the cardia may stimulate vagally mediated receptors, causing transient lower oesophageal sphincter relaxation and increasing the gastro-oesophageal reflux.(15)

Fig. 1: Various risk factors for gastro-oesophageal reflux disease
Has the negative relationship between H. pylori and gastro-oesophageal reflux disease been confirmed?
Epidemiological data on the frequency of gastro-oesophageal reflux disease, Barrett’s oesophagus, oesophageal adenocarcinoma and H. pylori
Figure 2 depicts the frequency of gastro-oesophageal reflux disease, and its sequelae such as Barrett’s oesophagus, oesophageal adenocarcinoma and H. pylori infection in various geographical areas of the world. It shows a high frequency of gastro-oesophageal reflux disease in developed countries like North America, Western Europe, Australia and Japan (19.4%, 10-21%, 17.5% and 17.9%, respectively).16-20 Corresponding high frequencies of Barrett’s oesophagus and oesophageal adenocarcinoma have been reported in North America and Western Europe (BE: 5-12%, 1-4% respectively; oesophageal adenocarcinoma: 5 per 100,000 population and 2.8-8.7 per 100,000, respectively).(21,22,23) The frequency of Barrett’s oesophagus in Australia and Japan is 0.3-1.9% and 0.9-1.2%, respectively and of oesophageal adenocarcinoma 4.8/100,000 and 6.9/100,000, respectively.(21,23,24) Conversely, South American, African, Eastern European, and Asian countries such as China and India have a lower frequency of gastro-oesophageal reflux disease and its sequel than the formerly mentioned developed nations (as shown in Figure 1).(25,26,27,28,29,30,31,32,33,34) This may be related to a difference in the frequency of H. pylori infection in these countries. While the frequency of H. pylori infection is lower in the Northern American, Western European, Australian and young Japanese populations (32%, 43%, 38% and 54.6%, respectively),(35,36,37,38) its frequency is higher in South Americans, Eastern Europeans, Africans and Chinese and Indians (76%, 70-92%, 61-100%, 44-60.4% and 81%, respectively).(26,38,39,40,41) This suggests that the presence of H. pylori may protect against gastro-oesophageal reflux disease and its complications.
In India, more than 80% of the adult population is infected with H. pylori.(42) There is a dearth of studies on the prevalence of gastro-oesophageal reflux disease and Barrett’s oesophagus in this population. However, a questionnaire-based study showed that 10.6% of healthy Indians in a community suffer symptoms of heartburn.(34) Another study showed that the incidence of oesophageal cancer in Indian men is between 3.5-6.7 per 100,000 individuals and in women is between 2.1-4.2 per 100,000 individuals (data extracted from the Cancer Registry, Indian Council of Medical Research).(43) As about 8% of all oesophageal cancers is oesophageal adenocarcinoma, in India,(44) we extrapolated the above data and thus concluded that 0.28-0.54 men per 100,000 and 0.17-0.34 women per 100,000 may develop oesophageal adenocarcinoma. Thus, the incidence of oesophageal adenocarcinoma in India is much lower than in the West. There is a single study from India evaluating the frequency of H. pylori in 30 patients with gastro-oesophageal reflux disease when compared with 30 patients of non-ulcer dyspepsia. Authors noted a comparable frequency of H. pylori infection in the two groups. However, the power of this study was very low and it is likely that the study had type II statistical errors due to the small sample size.(45) Most studies suggest that the severity and complications associated with gastro-oesophageal reflux disease such as peptic stricture, Barrett’s oesophagus and oesophageal adenocarcinoma may be lesser in the Indian population than in developed countries.(46,47) Thus, epidemiological data suggests a negative association between H. pylori and gastro-oesophageal reflux disease.

Fig. 2: World map showing frequency of gastro-oesophageal reflux disease, Barrett’s oesophagus, oesophageal adenocarcinoma and Helicobacter
pylori in community studies (shown within parenthesis). Frequency of gastro-oesophageal reflux disease, Barrett’s oesophagus and H. pylori are
given in percentage form whereas frequency of oesophageal adenocarcinoma is given in per 100,000 individuals. The respective references
are shown in superscript *: Endoscopic studies; **: prevalence of heartburn in general population; (GERD: gastro-oesophageal reflux disease;
BE: Barrett’s oesophagus; EAC: Oesophageal adenocarcinoma; Hp: Helicobacter pylori; ?: unknown; <: less than; %: percentage).
H. pylori infection influences gastric acid secretion profile
H. pylori may affect gastric acid secretion in two ways; (a) limited inflammation of the stomach due to localised infection at the antrum is associated with destruction of somatostatin secreting D-cells.(48) Thus, there is absence of the negative feedback effect on gastric acid secretion, leading to increased parietal cell mass and hypersecretion of acid thereby resulting in more severe gastro-oesophageal reflux disease, (b) severe inflammation of the entire stomach or pangastritis leads to destruction of acid secreting parietal cells of the body, causing hypo- or achlorhydria and consequent gastric atrophy, which may be associated with reduced severity of gastro-oesophageal reflux disease and its complications (Figure 3). Such variation in the pattern of gastritis may be related to host factors as has been reported in relation to another H. pylori associated disease, gastric adenocarcinoma.(49)
Available data indicate that the frequency of CagA-bearing strains of H. pylori (virulence factor of the bacterium) is lower in patients with complications of gastro-oesophageal reflux disease i.e. Barrett’s oesophagus and oesophageal adenocarcinoma in North Americans and Western Europeans.(14, 50,51) These data suggest that the presence of H. pylori, particularly of the CagA-bearing strains may protect against complications of gastro-oesophageal reflux disease. A study from Australia suggested that patients of Barrett’s oesophagus had a lower prevalence of H. pylori infection (p = 0.01) (52) indicating that this infection may have a protective role against oesophageal complications.
As the prevalence of Cag A bearing strains is higher in developing countries, including India,(53) individuals infected with these strains may develop corpus- or pangastritis leading to reduced gastric acid secretion and thus protecting against the development of gastro-oesophageal reflux disease.
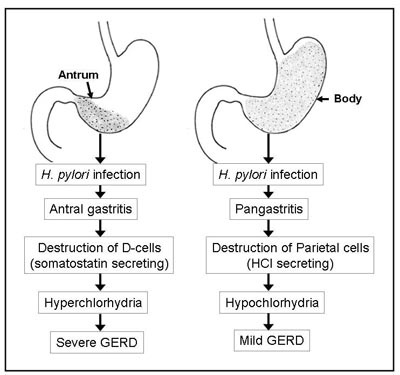
Fig. 3: Various outcomes of H. pylori infection and its relationship with gastric acid
secretion and gastro-oesophageal reflux disease.
Ethnic or inter-individual variations in response to H. pylori infection
The normal physiologic control of gastric secretory function and the effects of H. pylori infection on acid secretion depend to a certain extent on the genetic control of the inflammatory response.(54) At first contact with the host, lipopolysaccharide (LPS) present on the surface of H. pylori activates cells of the host innate immune system. The host defense systems are mobilised to eliminate the infection. LPS of many H. pylori strains express Lewis antigens (Lex, Ley, Lea, Leb), which are similar to the antigens expressed by gastric epithelial cells of the host. As, Lewis antigens are involved in cell surface adhesion and colonisation; the increased adherence may lead to increased bacterial load. This enhances the cross talk between H. pylori and the host and leads to activation of the transcription factor NF-êB and host signal transduction pathways.(54) In the stomach, locally produced IL-1â is an important mediator for the development of hypochlorhydria, since IL-1â inhibits gastric acid and pepsinogen secretion.(55) In healthy individuals, significant inter-individual variation in the in vitro production of pro-inflammatory proteins has been found.(56) Thus, host genetic make-up also plays a crucial role in the pathogenesis of disease.(49,57,58) However, little is known about the effect of genetic factors on gastro-oesophageal reflux disease pathogenesis.
Studies on the role of host genetic factors in the pathogenesis of gastro-oesophageal reflux disease and its sequelae
Figure 2 shows that there is variation in the prevalence of gastro-oesophageal reflux disease and its sequelae in different parts of the world. It also shows that there is marked variation in the frequency of H. pylori in different geographical areas. Therefore, it is possible that the differences in frequency, severity and complications of gastro-oesophageal reflux disease may be related either to the difference in frequency of H. pylori infection and/or genetic or racial factors. A susceptible individual may have a different genetic makeup to the resistant individual. Recently, it has been shown that certain genetic polymorphisms are associated with either decreased or increased risk of developing gastro-oesophageal reflux disease and its complications (Table 1).
CYP2C19 polymorphism
Although all proton pump inhibitors (PPIs) are very effective, the antisecretory effects of different drugs in this group vary among patients. This variation may be due to the acid-suppressing effect of H. pylori infection, which may augment the actions of the PPI. Secondly, this inter-patient variability in the effects of PPIs on acid secretion may be due to the involvement of genetically determined differences in the metabolism of these drugs. Human cytochrome P450 (CYP) enzymes, phase-I enzymes, play a key role in the metabolism of drugs and environmental chemicals. PPIs are eliminated by the hepatic route and the polymorphic CYP2C19 is involved in their metabolism. Three phenotypes have been identified in various populations: (i) extensive metabolisers (homEM), (ii) poor metabolisers (PM), and (iii) presence of one wild type and one mutant allele (hetEM). The PPI-induced healing rate in gastro-oesophageal reflux disease is apparently higher in PM/hetEM than in homEM of CYP2C19.59 For most PPIs, the activity of CYP2C19 determines the level of drug exposure, pharmacodynamic response (elevation of intragastric pH and serum levels of gastrin) and clinical outcome (H. pylori eradication, the healing rate of peptic ulcer and gastro-oesophageal reflux disease). The metabolism of PPIs such as omeprazole and lansoprazole, depends on the CYP2C19 genotype.60-62 Thus, testing for the CYP2C19 genotype may be potentially useful in managing gastro-oesophageal reflux disease with these drugs. On the other hand, rabeprazole does not undergo hepatic biotransformation by CYP2C19 and thus offers a significant advantage over the other PPIs as it is not affected by cytochrome polymorphis(63, 64 )

GST polymorphism
Glutathione S-transferase (GST) is an important phase-II enzyme of xenobiotic metabolism and carcinogen detoxification system of the gastrointestinal tract. GSTM1, GSTT1 and GSTP1 of this gene family exhibit polymorphism and may be associated with oesophageal(65) and gastric cancers.(66) Further studies have shown that GSTP1 genetic polymorphism may be one of the high susceptibility factors involved in the pathogenesis of gastro-oesophageal reflux disease. Further, patients with the positive variant of the GSTP1 genotype and who are negative for H. pylori infection have an increasing tendency to develop gastro-oesophageal reflux disease.(67) However, GSTM1 and GSTT1 genes did not show any relationship with gastro-oesophageal reflux disease in this study. Another study showed that the GSTP1b allele leads to lower GST enzyme activity and impaired detoxification in Barrett’s oesophagus and oesophageal adenocarcinoma.(68) However, a recent study also failed to show any association of GSTP1, GSTM1 and GSTT1 genes in gastro-oesophageal reflux disease, Barrett’s oesophagus and oesophageal adenocarcinoma. But, this study did show that variants of GSTT1 and GSTM1 genes are significantly higher in patients with oesophageal adenocarcinoma with a history of heavy smoking.(69)
IL-1B and IL-1RN gene polymorphism
Certain cytokines, such as IL-1â and IL-1Ra are potent mediators of inflammatory response and an imbalance between IL-1 and IL-1Ra production may lead to chronic inflammation.(70) Genetic polymorphisms in these inflammatory cytokines may have a particular impact on the inter-individual differences and pathogenesis of gastro-oesophageal reflux disease. Recent reports have suggested that certain proinflammatory gene polymorphisms have been associated with gastro-oesophageal reflux disease. IL1b-31 polymorphic alleles and IL1RN 2 alleles were shown to be negatively associated with gastro-oesophageal reflux disease.(71) IL1RN +2018 genotype 2/2 is associated with Barrett’s oesophagus more commonly than with gastro-oesophageal reflux disease.(72) However, subsequent studies suggest that there is no difference in the IL1b-511 polymorphism between gastro-oesophageal reflux disease, Barrett’s oesophagus and oesophageal adenocarcinoma.(72) This study however, did not compare this polymorphism with a healthy control population.
IL-10 polymorphism
This anti-inflammatory cytokine polymorphism at -1082 position (2/2 genotype) is more strongly associated with Barrett’s oesophagus and oesophageal adenocarcinoma than is gastro-oesophageal reflux disease.(72)
CCND1 polymorphism
Cyclin D1 encodes for a major cell cycle regulatory protein. It exhibits a polymorphism in exon 4 at G870A. A/A genotype has been associated with increased risk for gastro-oesophageal reflux disease, Barrett’s oesophagus, and oesophageal adenocarcinoma.(73)
DNA repair gene polymorphism
Various polymorphisms in DNA repair genes may be associated with gastro-oesophageal reflux disease and its sequelae. Oesophageal adenocarcinoma patients showed a higher frequency of XPC PAT (a nucleotide excision repair gene) homozygous variant than asymptomatic individuals, leading to increased risk.(74) Oesophageal adenocarcinoma also showed reduced frequency for the XPD (a nucleotide excision repair gene) Lys751Gln homozygous variant genotype, leading to reduced risk. Also, Barrett’s oesophagus and gastro-oesophageal reflux disease are associated with reduced frequency of XRCC1 (a base excision repair gene) Arg399Gln homozygous variant genotype, thus reducing the risk.(74) This study concluded that the malignant phenotype probably results from a summation of polymorphic nucleotide excision repair genes showing opposing effects. The protective effect of the XRCC1 for gastro-oesophageal reflux disease and Barrett’s oesophagus indicates that base excision repair alterations may occur early in the development of oesophageal adenocarcinoma.
Possible interactions in the final outcome of disease
Interplay between various environmental, dietary and host genetic factors determines the final outcome of a disease and this should be true for gastro-oesophageal reflux disease as well. Review of previous data on various risk factors for pathogenesis of gastro-oesophageal reflux disease reveals their potential role in the disease. Thus, gastro-oesophageal reflux disease is a multifactorial disease and many factors together influence the outcome.
Conclusions and future directions
Currently available data indicate a possible negative association between gastro-oesophageal reflux disease and H. pylori. This is supported by epidemiological data which suggest that the frequency of gastro-oesophageal reflux disease, Barrett’s oesophagus and oesophageal adenocarcinoma are higher in people in developed nations such as North America, Western Europe, Australia and Japan than in the developing world. Conversely, these developed nations have a lower frequency of H. pylori infection. Another possible reason for the low prevalence and severity of gastro-oesophageal reflux disease in developing nations may be related to the presence of more virulent strains of H. pylori (CagA) leading to corpus-/pangastritis, thus reducing gastric acid secretion resulting in decreased symptoms of gastro-oesophageal reflux disease. Further, ethnic differences or inter-individual variation also determine the disease outcome. This is mainly influenced by host genetic factors. Scarce data available have shown that homEM of CYP2C19, b allele (val105) of GSTP1, T allele of IL1B-31, 2/2 genotype of IL1RN +2018, 2/2 genotype of IL-10 -1082, A/A genotype of CCND1 G870A, and homozygous variant of XPC PAT gene are the potential risk factors for either gastro-oesophageal reflux disease or its complications (Barrett’s oesophagus and oesophageal adenocarcinoma).
Thus, we should not overlook the possibility of several risk factors being involved in the pathogenesis of gastro-oesophageal reflux disease as it may be a combination that determines disease outcome. This review shows a possible negative association between H. pylori and gastro-oesophageal reflux disease and its sequelae and throws light on the role of various host genetic factors which may aggravate or reduce the severity of gastro-oesophageal reflux disease. Our review of preliminary data on genetic factors should encourage further study on this issue, which may lead to better understanding of the disease and its treatment.
References
1. el-Serag HB, Sonnenberg A. Opposing time trends of peptic ulcer and reflux disease. Gut. 1998;43:327–33.
2. Falk GW. GERD and H. pylori: is there a link? Semin Gastrointest Dis. 2001;12:16–25.
3. Ahmed KS, Khan AA, Ahmed I, Tiwari SK, Habeeb A, Ahi JD, et al. Impact of household hygiene and water source on the prevalence and transmission of Helicobacter pylori: a South Indian perspective. Singapore Med J. 2007;48:543–9.
4. Holtmann G, Adam B, Liebregts T. Review article: the patient with gastro-oesophageal reflux disease—lifestyle advice and medication. Aliment Pharmacol Ther. 2004;20 Suppl 8:24–7.
5. Terry P, Lagergren J, Wolk A, Nyren O. Reflux-inducing dietary factors and risk of adenocarcinoma of the esophagus and gastric cardia. Nutr Cancer. 2000;38:186–91.
6. Gisbert JP, Pajares JM, Losa C. Helicobacter pylori and gastroesophageal reflux disease: friends or foes? Hepatogastroenterology. 1999;46:1023–9.
7. Vakil NB. Review article: gastro-oesophageal reflux disease and Helicobacter pylori infection. Aliment Pharmacol Ther. 2002;16 Suppl 1:47–51.
8. Richter J. Do we know the cause of reflux disease? Eur J Gastroenterol Hepatol. 1999;11 Suppl 1:S3–9.
9. Wu JC, Sung JJ, Ng EK, Go MY, Chan WB, Chan FK, et al. Prevalence and distribution of Helicobacter pylori in gastroesophageal reflux disease: a study from the East. Am J Gastroenterol. 1999;94:1790–4.
10. Fallone CA, Barkun AN, Friedman G, Mayrand S, Loo V, Beech R, et al. Is Helicobacter pylori eradication associated with gastroesophageal reflux disease? Am J Gastroenterol. 2000;95:914–20.
11. Haruma K. Review article: influence of Helicobacter pylori on gastro-oesophageal reflux disease in Japan. Aliment Pharmacol Ther. 2004;20 Suppl 8:40–4.
12. Vicari J, Falk GW, Richter JE. Helicobacter pylori and acid peptic disorders of the esophagus: is it conceivable? Am J Gastroenterol. 1997;92:1097–102.
13. el-Omar EM, Penman ID, Ardill JE, Chittajallu RS, Howie C, McColl KE. Helicobacter pylori infection and abnormalities of acid secretion in patients with duodenal ulcer disease. Gastroenterology. 1995;109:681–91.
14. Vicari JJ, Peek RM, Falk GW, Goldblum JR, Easley KA, Schnell J, et al. The seroprevalence of cagA-positive Helicobacter pylori strains in the spectrum of gastroesophageal reflux disease. Gastroenterology. 1998;115:50–7.
15. Zerbib F, Bicheler V, Leray V, Joubert M, Bruley des Varannes S, Galmiche JP. H. pylori and transient lower esophageal sphincter relaxations induced by gastric distension in healthy humans. Am J Physiol Gastrointest Liver Physiol. 2001;281:G350–6.
16. Nastaskin I, Mehdikhani E, Conklin J, Park S, Pimentel M. Studying the overlap between IBS and GERD: a systematic review of the literature. Dig Dis Sci. 2006;51:2113–20.
17. Bollschweiler E, Knoppe K, Wolfgarten E, Holscher AH. [Prevalence of reflux symptoms in the general population of Cologne]. Z Gastroenterol. 2007;45:177–81.
18. Mohammed I, Nightingale P, Trudgill NJ. Risk factors for gastro-oesophageal reflux disease symptoms: a community study. Aliment Pharmacol Ther. 2005;21:821–7.
19. Talley NJ, Boyce P, Jones M. Identification of distinct upper and lower gastrointestinal symptom groupings in an urban population. Gut. 1998;42:690–5.
20. Mishima I, Adachi K, Arima N, Amano K, Takashima T, Moritani M, et al. Prevalence of endoscopically negative and positive gastroesophageal reflux disease in the Japanese. Scand J Gastroenterol. 2005;40:1005–9.
21. Hongo M. Review article: Barrett’s oesophagus and carcinoma in Japan. Aliment Pharmacol Ther. 2004;20 Suppl 8:50–4.
22. Celinski K, Slomka M, Kasztelan-Szczerbinska B, Madro A, Cichoz-Lach H, Kurzeja A. Helicobacter pylori infection and the risk of adenocarcinoma of the esophagus. Ann Univ Mariae Curie Sklodowska [Med]. 2004;59:424–7.
23. Bollschweiler E, Wolfgarten E, Gutschow C, Holscher AH. Demographic variations in the rising incidence of esophageal adenocarcinoma in white males. Cancer. 2001;92:549–55.
24. Kendall BJ, Whiteman DC. Temporal changes in the endoscopic frequency of new cases of Barrett’s esophagus in an Australian health region. Am J Gastroenterol. 2006;101:1178–82.
25. Chiocca JC, Olmos JA, Salis GB, Soifer LO, Higa R, Marcolongo M. Prevalence, clinical spectrum and a typical symptoms of gastro-oesophageal reflux in Argentina: a nationwide population-based study. Aliment Pharmacol Ther. 2005;22:331–42.
26. Segal I. The gastro-oesophageal reflux disease complex in sub-Saharan Africa. Eur J Cancer Prev. 2001;10:209–12.
27. Papatheodoridis GV, Karamanolis DG. Prevalence and impact of upper and lower gastrointestinal symptoms in the Greek urban general population. Scand J Gastroenterol. 2005;40:412–21.
28. Wong WM, Lai KC, Lam KF, Hui WM, Hu WH, Lam CL, et al. Prevalence, clinical spectrum and health care utilization of gastro-oesophageal reflux disease in a Chinese population: a population-based study. Aliment Pharmacol Ther. 2003;18:595–604.
29. Andreollo NA, Michelino MU, Brandalise NA, Lopes LR, Trevisan MA, Leonardi LS. [Incidence and epidemiology of Barrett’s epithelium at the Gastrocentro-UNICAMP]. Arq Gastroenterol. 1997;34:22–6.
30. Wong WM, Lam SK, Hui WM, Lai KC, Chan CK, Hu WH, et al. Long-term prospective follow-up of endoscopic oesophagitis in southern Chinese—prevalence and spectrum of the disease. Aliment Pharmacol Ther. 2002;16:2037–42.
31. el-Serag HB. The epidemic of esophageal adenocarcinoma. Gastroenterol Clin North Am. 2002;31:421–40.
32. Botterweck AA, Schouten LJ, Volovics A, Dorant E, van Den Brandt PA. Trends in incidence of adenocarcinoma of the oesophagus and gastric cardia in ten European countries. Int J Epidemiol. 2000;29:645–54.
33. Fernandes ML, Seow A, Chan YH, Ho KY. Opposing trends in incidence of esophageal squamous cell carcinoma and adenocarcinoma in a multi-ethnic Asian country. Am J Gastroenterol. 2006;101:1430–6.
34. Shah SS, Bhatia SJ, Mistry FP. Epidemiology of dyspepsia in the general population in Mumbai. Indian J Gastroenterol. 2001;20:103–6.
35. Simon JA, Hudes ES, Perez-Perez GI. Relation of serum ascorbic acid to Helicobacter pylori serology in US adults: the Third National Health and Nutrition Examination Survey. J Am Coll Nutr. 2003;22:283–9.
36. Seery JP, Henshaw DJ, Sandhu PJ, Mather HM, Ahmad F, McNeil I, et al. Helicobacter pylori infection and upper gastrointestinal patholgy in a British immigrant Indian community. Eur J Gastroenterol Hepatol. 1997;9:191–4.
37. Lin SK, Lambert JR, Nicholson L, Lukito W, Wahlqvist M. Prevalence of Helicobacter pylori in a representative Anglo-Celtic population of urban Melbourne. J Gastroenterol Hepatol. 1998;13:505–10.
38. Singh K, Ghoshal UC. Causal role of Helicobacter pylori infection in gastric cancer: an Asian enigma. World J Gastroenterol. 2006;12:1346–51.
39. Mattana C, Vega A, Gomez P, Puig de Centorbi O. [Serological profile of Helicobacter pylori infection in the population of San Luis (Argentina)]. Enferm Infecc Microbiol Clin. 2004;22:227–9.
40. Reshetnikov OV, Haiva VM, Granberg C, Kurilovich SA, Babin VP. Seroprevalence of Helicobacter pylori infection in Siberia. Helicobacter. 2001;6:331–6.
41. Jun ZJ, Lei Y, Shimizu Y, Dobashi K, Mori M. High seroprevalence of Helicobacter pylori in chronic bronchitis among Chinese population. The Tohoku J Exp Med. 2006;208:327–331.
42. Mazumder DN, Ghoshal UC. Epidemiology of Helicobacter pylori in India. Indian J Gastroenterol. 1997;16 Suppl 1:S3–5.
43. Chaudhry K, Luthra UK. 50 Years of Cancer Control in India: CANCER REGISTRATION IN INDIA. Indian Council of Medical Research.14–26.
44. Cherian JV, Sivaraman R, Muthusamy AK, Jayanthi V. Carcinoma of the esophagus in Tamil Nadu (South India): 16-year trends from a tertiary center. J Gastrointestin Liver Dis. 2007;16:245–9.
45. Yerra LN, Bhasin DK, Panigrahi D, Vaiphei K, Sharma BC, Ray P. Prevalence of Helicobacter pylori infection in patients with reflux oesophagitis. Trop Gastroenterol. 1999;20:175–7.
46. Saraswat VA, Dhiman RK, Mishra A, Naik SR. Correlation of 24-hr esophageal pH patterns with clinical features and endoscopy in gastroesophageal reflux disease. Dig Dis Sci. 1994;39:199–205.
47. Somani SK, Ghoshal UC, Saraswat VA, Aggarwal R, Misra A, Krishnani N, et al. Correlation of esophageal pH and motor abnormalities with endoscopic severity of reflux esophagitis. Dis Esophagus. 2004;17:58–62.
48. Kamada T, Haruma K, Kawaguchi H, Yoshihara M, Sumii K, Kajiyama G. The association between antral G and D cells and mucosal inflammation, atrophy, and Helicobacter pylori infection in subjects with normal mucosa, chronic gastritis, and duodenal ulcer. Am J Gastroenterol. 1998;93:748–52.
49. Ghoshal UC, Tripathi S, Ghoshal U. The Indian enigma of frequent H. pylori infection but infrequent gastric cancer: is the magic key in Indian diet, host’s genetic make up, or friendly bug? Am J Gastroenterol. 2007;102:2113–4.
50. Vaezi MF, Falk GW, Peek RM, Vicari JJ, Goldblum JR, Perez-Perez GI, et al. CagA-positive strains of Helicobacter pylori may protect against Barrett’s esophagus. Am J Gastroenterol. 2000;95:2206–11.
51. Ackermark P, Kuipers EJ, Wolf C, Breumelhof R, Seldenrijk CA, Timmer R, et al. Colonization with cagA-positive Helicobacter pylori strains in intestinal metaplasia of the esophagus and the esophagogastric junction. Am J Gastroenterol. 2003;98:1719–24.
52. Lord RV, Frommer DJ, Inder S, Tran D, Ward RL. Prevalence of Helicobacter pylori infection in 160 patients with Barrett’s oesophagus or Barrett’s adenocarcinoma. Aust N Z J Surg. 2000;70:26–33.
53. Ghoshal UC, Tiwari S, Pandey Rea. Frequency of Helicobacter 53. Ghoshal UC, Tiwari S, Pandey Rea. Frequency of Helicobacter pylori and CagA antibody in patients with gastric neoplasm and controls: The Indian enigma. Am J Gastroneterol. 2005;100 (suppl):A64.
54. Pena A. Genetic factors determining the host response to Helicobacter pylori. World J Gastroenterol. 2000;6:624–625.
55. Liou JM, Lin JT, Wang HP, Huang SP, Lee YC, Chiu HM, et al. IL-1B-511 C—>T polymorphism is associated with increased host susceptibility to Helicobacter pylori infection in Chinese. Helicobacter. 2007;12:142–9.
56. Yaqoob P, Newsholme EA, Calder PC. Comparison of cytokine production in cultures of whole human blood and purified mononuclear cells. Cytokine. 1999;11:600–5.
57. van Deventer SJ. Cytokine and cytokine receptor polymorphisms in infectious disease. Intensive Care Med. 2000;26 Suppl 1:S98–102.
58. Cullup H, Stark G. Interleukin-1 polymorphisms and graft-vs-host disease. Leuk Lymphoma. 2005;46:517–23.
59. Klotz U, Schwab M, Treiber G. CYP2C19 polymorphism and proton pump inhibitors. Basic Clin Pharmacol Toxicol. 2004;95:2–8.
60. Sagar M, Bertilsson L, Stridsberg M, Kjellin A, Mardh S, Seensalu R. Omeprazole and CYP2C19 polymorphism: effects of long-term treatment on gastrin, pepsinogen I, and chromogranin A in patients with acid related disorders. Aliment Pharmacol Ther. 2000;14:1495–502.
61. Kawamura M, Ohara S, Koike T, Iijima K, Suzuki J, Kayaba S, et al. The effects of lansoprazole on erosive reflux oesophagitis are influenced by CYP2C19 polymorphism. Aliment Pharmacol Ther. 2003;17:965–73.
62. Kawamura M, Ohara S, Koike T, Iijima K, Suzuki H, Kayaba S, et al. Cytochrome P450 2C19 polymorphism influences the preventive effect of lansoprazole on the recurrence of erosive reflux esophagitis. J Gastroenterol Hepatol. 2007;22:222–6.
63. Horn J. Review article: relationship between the metabolism and efficacy of proton pump inhibitors—focus on rabeprazole. Aliment Pharmacol Ther. 2004;20 Suppl 6:11–9.
64. Kinoshita Y. Review article: treatment for gastro-oesophageal reflux disease—lifestyle advice and medication. Aliment Pharmacol Ther. 2004;20 Suppl 8:19–23.
65. Casson AG, Zheng Z, Porter GA, Guernsey DL. Genetic polymorphisms of microsomal epoxide hydroxylase and glutathione S-transferases M1, T1 and P1, interactions with smoking, and risk for esophageal (Barrett) adenocarcinoma. Cancer Detect Prev. 2006;30:423–31.
66. Tripathi S, Ghoshal U, Ghoshal UC, Mittal B, Krishnani N, Agarwal A, et al. Gastric carcinogenesis: Possible role of polymorphisms of GSTM1, GSTT1 and GSTP1 genes. Scand J Gastroenterol;(in press).
67. Liu B, Fan YJ, Wang ML, Lu XD, Jiang JL, Wang TY, et al. Genetic polymorphisms in glutathione S-transferases T1, M1 and P1 and susceptibility to reflux esophagitis. Dis Esophagus. 2006;19:477–81.
68. van Lieshout EM, Roelofs HM, Dekker S, Mulder CJ, Wobbes T, Jansen JB, et al. Polymorphic expression of the glutathione S-transferase P1 gene and its susceptibility to Barrett’s esophagus and esophageal carcinoma. Cancer Res. 1999;59:586–9.
69. Kala Z, Dolina J, Marek F, Izakovicova Holla L. Polymorphisms of glutathione S-transferase M1, T1 and P1 in patients with reflux esophagitis and Barrett’s esophagus. J Hum Genet. 2007;52:527–34.
70. Casini-Raggi V, Kam L, Chong YJ, Fiocchi C, Pizarro TT, Cominelli F. Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease. A novel mechanism of chronic intestinal inflammation. J Immunol. 1995;154:2434–40.
71. Queiroz DM, Guerra JB, Rocha GA, Rocha AM, Santos A, De Oliveira AG, et al. IL1B and IL1RN polymorphic genes and Helicobacter pylori cagA strains decrease the risk of reflux esophagitis. Gastroenterology. 2004;127:73–9.
72. Gough MD, Ackroyd R, Majeed AW, Bird NC. Prediction of malignant potential in reflux disease: are cytokine polymorphisms important? Am J Gastroenterol. 2005;100:1012–8.
73. Casson AG, Zheng Z, Evans SC, Geldenhuys L, van Zanten SV, Veugelers PJ, et al. Cyclin D1 polymorphism (G870A) and risk for esophageal adenocarcinoma. Cancer. 2005;104:730–9.
74. Casson AG, Zheng Z, Evans SC, Veugelers PJ, Porter GA, Guernsey DL. Polymorphisms in DNA repair genes in the molecular pathogenesis of esophageal (Barrett) adenocarcinoma. Carcinogenesis. 2005;26:1536–41.