48uep6bbphidvals|249
48uep6bbphidcol4|ID
48uep6bbph|2000F98CTab_Articles|Fulltext
Desmoplastic Small Round Cell Tumor (DSRCT) is a rare aggressive malignancy seen in male patients in their 2nd or 3rd decade of life.[1,2,3,4,5,6] These tumors mainly occur in the peritoneal cavity, although other primary sites such as paratesticular[7], intracranial[8], thoracic[9] have been reported. Approximately 200 cases have been reported in literature[10]. Common presenting symptoms and signs are abdominal ain, palpable mass etc.[5,11,12] These tumours have a distinctive nesting pattern of cellular growth with dense desmoplastic stroma and immunohistochemical co-expression of epithelial, uscle and neural markers. [1,2,13] The chromosomal abnormality is t(11;22) (p13;q12)[14] and the break points involve two genes associated specifically with Ewing’s sarcoma and Wilm’s tumor. Surgical resection with adjuvant chemotherapy may relieve symptoms and prolong survival in some patients.
Case Report
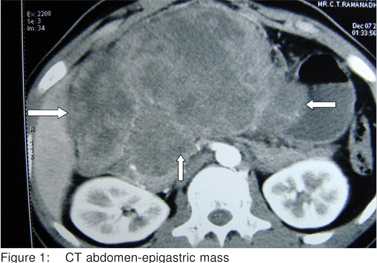
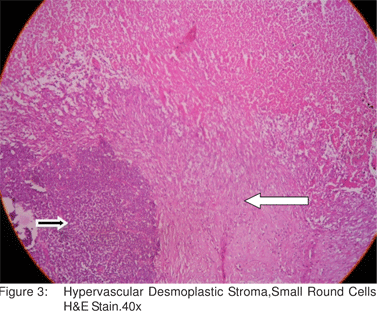
A 21 year old male presented with abdominal pain of 3 months duration and recent complaints of vomiting, loose stools and weight loss of 5 kgs in 3 weeks. On examination, he had a huge epigastric intra abdominal mass. Blood Investigations were within normal limits. CT Scan showed a large lobulated heterogenous mass lesion in epigastric region (17x8.7x6.3cm)(Figure 1). Endoscopy showed external compression of stomach and D1 and mucosa appeared normal. CT Angio showed a hypovascular mass supplied by celiac and superior mesenteric artery, with loss of fat plane between mass and main portal vein. Laparotomy was done which showed a huge mass (17x10x8cm) in the epigastric region adherent to the undersurface of liver, pylorus, D1 and greater omentum. Tumor was excised completely along with subtotal gatrectomy and partial duodenectomy of D1 (Figure 2). Histopathologic examination demonstrated sheets, nests and lobules of small round cells with hyperchromatic nuclei. The cells were surrounded by a desmoplastic stroma (Figure 3). The Immunohistochemical tests were positive for vimentin. Postoperatively, patient was started on adjuvant multi drug chemotherapy.
Discussion
DSRCT is a highly aggressive rare malignancy seen in young patients with male predominance. Symptoms are usually abdominal pain and abdominal mass. DSRCT is associated with poor prognosis with a mean survival of 16 to 30 months. In a study on CT findings in 11 patients[15], patients had multiple omental, serosal or mesenteric masses. Isolated dominant masses in the upper and mid abdomen were seen in only 2 patients. 9 patients had dominant masses in the retrovesical or rectouterine spaces (size varied between 6 to 25cms). All dominant tumors displayed heterogenous enhancement after IV contrast. Other findings include punctuate tumor calcification, lymphadenopathy, ascites and hypoattenuating liver metastases. In another study,[16] designed to correlate radiologic and histopathologic findings, the most characteristic finding was multiple peritoneal soft tissue masses without an apparent organ based primary site. The diameter varied between 2 to 12cms.The tumors were predominantly intraperitoneal with paravseical and omental sites each involved in 6 of 9 patients. Solitary peritoneal masses were located in the omentum and paravesical region.

Other findings included liver metastases, lymphadenopathy and ascites[16]. Histologic examination remains the gold standard for DSRCT ,aided by immunoctochemical and cytogenetic studies. Immunohistochemically, DRSCT demonstrates a divergent differentiation. Typically, tumor cells are immunoreactive for epithelial (keratin and epithelial membrane antigen), mesenchymal (vimentin), myogenic (desmin) and neural (neuron specific enolase and CD56)markers. Cytogenetic studies have demonstrated a characteristic reciprocal chromosomal translocation, t(11;22)(p13;q12), which is different from the t(11;22)(q24;q12) translocation observed in Ewing sarcoma/PNET .The chimeric transcript corresponding to the fusion gene product can be detected by reverse transcriptase-polymerase chain reaction(RT-PCR) technique. Histologically and cytologically, DSRCT must be distinguished from other small round cell tumors such as Ewing’s sarcoma/PNET, small cell carcinoma, lymphoma, neuroblastoma, Wilm’s tumor , rhabdomyosarcoma and malignant mesothelioma. Surgery along with chemotherapy has been the mainstay of treatment though there is no standardized treatment protocol. A study[17] using cisplatin, etoposide, cyclophosphamide, and either doxorubicin or epirubicin (PAVEP/PEVEP regimen was done in 5 patients. 4 patients (intraabdominal tumor) who received PAVEP showed stability ranging from 4 to 9 months, objective persistent complete response was observed only in one patient (paratesticular tumor) who received PEVEP. After 4 cycles of PEVEP, (Doxorubicin for Epirubicin), two courses of high dose Carboplatin, Etoposide and Ifosamide with stem cell support was given as consolidation therapy. That patient was alive 32 months after diagnosis with no signs of recurrence. One patient had complete remission lasting 42 months and was treated with abdominal radiation along with 5 FU.[17] In another study,[18] patients with DSRCT were treated with high dose alkylator based regimen labeled as P6 Protocol. 10 patients previously untreated and 2 patients previously treated were assessed. 7 out of 10 untreated patients had partial response or very good response to the protocol which included HD-CAV (high dose Cycloposphamide, Doxorubicin and Vincristine). 2 patients had no assessable disease after tumor resection at diagnosis. After chemotherapy and surgery, 7 patients were in complete remission and 2 were in partial remission. 5 out of 7 patients in complete remission remained event free.[18] A review of literature shows that 60 patients who were treated with chemotherapy with or without radiotherapy, an objective response was seen in 17 patients , 8 had complete response, 3 of 8 developed recurrence ,other 5 are in complete remission after 4,12,24,42 and 48 months. The regimen associated with complete response were those using Doxorubicin, Cycloposphamide, Vincristine and Cisplatin.[17] A study by Gil et al[12] reported that median survival was 20 months in 4 patients with complete resection compared with 11 months in 3 cases without complete resection.
Refrences
1. Geral WL,Rosai J. Case 2:desmolplastic small round cell tumor with divergent differentiation. Pediatr Pathol. 1989;9:177–83.
2. Geral WL, Miller HK, Battifora H, Miettinen M, Silva EG, Rosai J. Intra-abdominaldesmoplastic small round cell tumor:repot of 19 cases of a distinctive type of high grade polyphenotypic malignancy affecting young individuals. Am J Surg Pathol. 1991;15:499–513.
3. Dorsey BV,Benjamin LE,Rauscher F, Klencke B, Venook AP, Warren RS, et al. Intra-abdominal demoplastic small round-cell tumor:expansion of pathologic profile. Mod Pathol. 1996;9:703– 9.
4. Amato RJ, Ellerhorst JA, Ayala AG. Intraabdominal desmoplastic small round cell tumor. Report and discussion of five cases. Cancer. 1996;78:845–51.
5. Gerald WL,Ladanyi M,de Alava E.et al .Clinical,pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round cell tumor and its variants. J Clin Oncol. 1998;16:3028–36.
6. Lae ME, Roche PC, Jin L, Llyod RV, Nascimento AG. Desmoplastic small round cell tumor: a clinicopatologic, immmunohistochemical, and molecular study of 32 tumors. Am J Surg Pathol . 2002;26:823–35.
7. Cummings OW, Ulbright TM, Young RH, Dei Tos AP, Fletcher CD, Hull MT. Desmoplastic small round cell tumors of paratesticular region. A report of six cases. Am J Surg Pathol . 1997;21:219–25.
8. Tison V, Cerasoli S, Morigi F, Ladanyi M, Gerald WL, Rosai J. Intracranial desmoplastic small cell tumor. Report of a case. Am J Surg Pathol. 1996;20:112–7.
9. Syed S ,Haque AK, Hawkins HK, Soernsen PH, Cowan DF. Desmoplastic small round cell tumor of the lung. Arch Pathol Lab Med. 2002;126:1226–8.
10. Chang F. Desmoplastic small round cell tumors: cytologic, histologic and immunohistochemical features. Arch Pathol Lab Med. 2006;130:728–32.
11. Quaglia MP, Brennan MF. The clinical approach to desmoplastic small round cell tumor. Surg Oncol. 2000;9:77–81.
12. Gil A, Gomez Portilla A, Brun EA, Sugarbaker PH. Clinical perspective on desmoplastic small round-cell tumor. Oncology. 2004;67:231–42.
13. Ordonez NG, el-Naggar AK, Ro JY, Silva EG, Mackay B. Intraabdominal desmoplastic small cell tumor: a light microscopic, immunohistochemical , ultrastructural and flowcytometric study. Hum Pathol. 1993;24:850–65.
14. Biegel JA, Conard K, Brooks JJ. Translocation (11;22)(p13;q12): primary change in intra-abdominal desmoplastic small round cell tumor. Genes Chromosomes Cancer. 1993;7:119–21.
15. Bellah R, Suzuki-Bordalo L, Brecher E, Ginsberg JP, Maris J, Pawel BR. Desmoplastic small round cell tumor in the abdomen and pelvis: report of CT findings in 11 affected children and young adults. AJR. 2005;184:1910–4.
16. Pickhardt PJ, Fisher AJ, Balfe DM, Dehner LP, Heuttner PC. Desmoplastic small round cell tumor of the abdomen: radiologichistopathologic correlation. Radiology. 1999;210:633–8.
17. Farhat F, Culine S, Lhomme C, Duvillard P, Soulie P, Michel G, et al. Desmoplastic small round cell tumors: results of a four-drug chemotherapy regimen in five adult pat ients. Cancer . 1996;77:1363–6.
18. Kushner BH, LaQuaglia MP, Wollner N, Meyers PA, Lindsley KL, Ghavimi F, et al. Desmoplastic small round- cell tumor: prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol. 1996;14:1526–31.