48uep6bbphidcol2|ID
48uep6bbphidvals|1905
48uep6bbph|2000F98CTab_Articles|Fulltext
Acute viral hepatitis A is one of the common causes of acute hepatitis. Acute kidney injury (AKI) has been reported in 1.5% - 4.7% of non-fulminant hepatitis A.1 The mechanism of hepatitis A associated AKI is uncertain for which various explanations were described in literature.2 Hyperbilirubinemia was also described as a cause for renal failure.3 We came across a case of young male with acute hepatitis A complicated with oliguric AKI which was diagnosed as bile-cast nephropathy and recovered with longstanding renal replacement therapy.
Case Report
A previously healthy 22-year old male presented with a history of fever, jaundice, loose stools, and vomiting of 5 days duration. It was associated with pain in the right upper quadrant of the abdomen and decreased urine output. Examination revealed pallor and icterus but no features of hepatic encephalopathy or stigmata of chronic liver disease. His systemic examination was unremarkable except for tender hepatomegaly. His initial investigation reports revealed hemoglobin of 9.4 g/dl, total count 38,900 cells/mm3, total bilirubin 40 mg/dl with a direct fraction of 30 mg/dl, ALT 1853 IU/L, AST 2016 IU/L, ALP 189 IU/L, PT INR 1. Renal functions were deranged with a serum creatinine of 3.9 mg/dl, sodium 127 mmol/L, K- 5.5 mmol/L. IgM against Hepatitis A was positive. IgM anti-HEV, HBsAg and anti-HCV were negative. ABG revealed compensated metabolic acidosis. Workup for scrub typhus, malaria, dengue, leptospirosis, and autoimmune hepatitis was negative. Complement C3, C4, pANCA, and cANCA were normal. Features of hemolysis were noted on peripheral smear, with negative coomb’s test, reticulocyte count of 10%, haptoglobin 1.4 mg/dl, LDH 5760 IU/L. Ceruloplasmin levels were normal (30 mg/dL), and Kayser Fleischer ring was not detected. Serum ferritin levels were very high (39,700 ng/mL), serum iron 217 mcg/dL, TS-94% and TIBC-212 mcg/dL. Blood and urine cultures were sterile.
USG abdomen showed mild hepatosplenomegaly, GB wall edema with mild ascites. He was hydrated well and started on antibiotics and other supportive measures. On day 3, his urine output declined further, and serum creatinine increased to 7 mg/dl. Renal replacement therapy was initiated besides other supportive measures. On day 10, his total serum bilirubin decreased to 16 mg/dl, and transaminitis improved; However, his renal functions did not improve. Renal biopsy was performed, which revealed acute tubular necrosis with interstitial edema. Special stain showed bile casts within the tubular lumen along with features of tubular epithelial injury indicating bile cast nephropathy (Figure 1). After about 6 weeks of anuria and hemodialysis, his urine output gradually increased, renal parameters also declined slowly, and the need for dialysis decreased. At the end of 10 weeks, his creatinine improved to 1.6 mg/dl, bilirubin 1.1 mg/dl, AST/ALT 170/120, and his general condition improved to pre-morbid state.
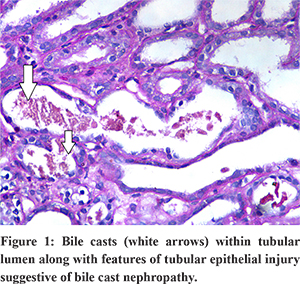
Discussion
The mechanism of hepatitis A associated AKI is uncertain, and several possible mechanisms have been offered. First, prerenal azotemia due to hypovolemia may progress to acute tubular necrosis and AKI. Second, immune complex-mediated nephritis is possible and glomerular disorders such as membranous nephropathy, mesangial proliferative glomerulonephritis, and membranoproliferative glomerulonephritis have been reported in HAV infected patients.2 Third, acute hepatitis-related endotoxemia may cause renal injury. Fourth, hyperbilirubinemia may cause vasoconstriction of the renal vasculature.3 A direct cytopathic effect of the virus has also been proposed as a cause of AKI.
Bile cast nephropathy (BCN) is a rare and poorly understood entity characterized by progressive renal dysfunction in the setting of elevated serum bile salts and hyperbilirubinemia. In 2006, Betjes and Bajima used the term “jaundice-related nephropathy” for the historical term cholemic nephropathy for changes ranging from proximal tubular dysfunction to renal failure due to the deposition of bile and bile salts.4 The excess bilirubin is believed to cause oxidative damages of the cell membranes of the tubules and uncoupling of mitochondrial phosphorylation at the cellular level.5,6 Furthermore, inhibition of the Na-H, Na-K, Na-Cl pumps by sulfated bile salts in proximal tubules and in the loop of Henle resulting in pH changes which enhance bile cast deposition and tubular injury.7 It has been hypothesized that there is a limit to bilirubin transport in the proximal tubules, after which they become saturated, leading to cast formation and tubular obstruction.8 The primary findings in bile cast nephropathy include renal tubular hypertrophy, the presence of pigmented bile casts within the renal tubules and absence of glomerular pathology. Nayak et al, found that patients with BCN had significantly higher levels of serum total bilirubin, total leukocyte count, and model for end-stage liver disease score, as compared to those without BCN.9 Further, a lower platelet count, presence of coagulopathy and an elevated AST or ALT on presentation were independent risk factors for AKI in patients with AHA.10
There are currently no accepted treatment guidelines owing to the rarity of this condition. Interventions to reduce bilirubin burden, such as hemodialysis and plasmapheresis, may be of utility. Extracorporeal treatment options have also emerged: including the molecular adsorbents recycling system (MARS), coupled plasma filtration adsorption, plasma filtration adsorption dialysis.
In conclusion, bile cast nephropathy is a rare entity that results from multiple concurrent insults to the kidney, including direct toxicity from bile acids, obstructive physiology from bile casts, and systemic hypoperfusion from vasodilation. Obtaining a renal biopsy should be considered in patients suspected of this entity to aid in the diagnosis.
References
- Lin CC, Chang CH, Lee SH, Chiang SS, Yang AH. Acute renal failure in non-fulminant hepatitis A. Nephrol Dial Transplant. 1996;11:2061–2066.
- Morita M, Kitajima K, Yoshizawa H, Itoh Y, Iwakiri S, Shibata C, Mayumi M. Glomerulonephritis associated with arteritis in marmosets infected with hepatitis A virus. Br J Exp Pathol. 1981;62:103–113.
- Green J, Beyar R, Bomzon L, Finberg JP, BetterOS: Jaundice, the circulation and the kidney. Nephron 1984; 37:145-152.
- Betjes MG, Bajema I. The pathology of jaundice-related renal insufficiency: cholemic nephrosis revisited. J Nephrol 2006;19:229-233.
- Wu B, Gong D, Ji D, Xu B, Liu Z. Clearance of myoglobin by high cutoff continuous veno-venous hemodialysis in a patient with rhabdomyolysis: a case report. Hemodial Int. 2015;19:135–140.
- Fickert P, Krones E, Pollheimer MJ, Thueringer A, Moustafa T, Silbert D, Halilbasic E, Yang M, Jaeschke H, Stokman G, et al. Bile acids trigger cholemic nephropathy in common bile-duct-ligated mice.Hepatology. 2013;58:2056–2069.
- Sellinger M, Haag K, Burckhardt G, Gerok W, Knauf H. Sulfatedbile acids inhibit Na(+)-H+ antiport in human kidney brush-border membrane vesicles. Am J Physiol 1990; 258: F986-F991.
- Van Slambrouck CM, Salem F, Meehan SM, Chang A. Bile cast nephropathy is a common pathologic finding for kidney injury associated with severe liver dysfunction. Kidney Int 2013; 84:192-197.
- Nayak SL, Kumar M, Bihari C, Rastogi A. Bile Cast Nephropathy in Patients with Acute Kidney Injury Due to Hepatorenal Syndrome: A Postmortem Kidney Biopsy Study. Journal of Clinical and Translational Hepatology. 2017;5(2):92-100.
- Shin SJ, Kim JH. The characteristics of acute kidney injury complicated in acute hepatitis A. Scand J Infect Dis. 2009;41(11-12):869-72.