48uep6bbphidvals|487
48uep6bbph|2000F98CTab_Articles|Fulltext
Physiologic jaundice occurs in 65% of full-term infants. However, persistent hyperbilirubinemia beyond 2 weeks should raise concern for several other possibilities requiring immediate intervention. Craig and Landing in 1952 first distinguished neonatal hepatitis from extrahepatic biliary atresia.[1] Intrahepatic biliary atresia was recognized later. The etiology of all 3 conditions remains ill defined. Neonatal hepatitis and congenital biliary atresia account for the vast majority of persistent jaundice beyond 2 weeks in a neonate.
Biliary atresia was initially thought to be a congenital malformation. The liver and biliary system develops from a bi-lobed endodermal bud. The cephalic bud gives rise to the right and left lobes of the liver. The caudal bud gives rise to biliary tract and the gallbladder. The biliary tract proper is formed after recanalization of the solid core by 12–14 weeks of intrauterine life.[2] Bile secretion begins thereafter and flows into the small intestine. Most of the congenital abnormalities of the biliary tract are thought to be due to either failure of recanalization of the ducts (biliary atresia) or faulty recanalization with cystic dilatation of intrahepatic (Caroli’s disease) and extrahepatic ducts (choledochal cyst).
Subsequent research and clinical evidence did not support the hypothesis of congenital malformation. Now it is believed that biliary atresia is a progressive inflammatory panductular obliterative disease of the bile ducts starting in the antenatal period. The destructive inflammatory process might involve only the distal part of the extrahepatic bile duct causing obstruction and leaving the proximal ducts patent. The clinical presentation is that of infantile obstructive cholangiopathy, waxing and waning icterus, pale colored stools and high colored urine from early neonatal period. Nearly 90% of biliary atresia is extrahepatic in location and 10% is intrahepatic, often associated with various syndromes namely Alagille syndrome, Trisomy 18, Trisomy 19, etc. As mentioned above extrahepatic biliary atresia (EHBA) is a progressive, sclerosing, inflammatory disease, primarily affecting the extrahepatic bile duct from the region of the porta hepatis to its termination in the duodenum. It is estimated to affect 1 in 13,000 live births and is more commonly seen in Asian and Oriental neonates.[3]
In our own series from AIIMS, the etiology of neonatal cholestasis in 132 kids was: 25% (33/132) EHBA, 45.5% (60/132) neonatal hepatitis (NH) with an identifiable cause and 29.5% (39/132) idiopathic NH.[4] The diagnosis is suggested by raised direct bilirubin level and pale stool, and confirmed by a per-operative cholangiogram. In liver biopsy, the histologic changes of biliary atresia are indistinguishable from neonatal hepatitis however, some changes have prognostic significance. Initial management is surgical (portoenterostomy or Kasai operation), entailing excision of the atretic biliary tree and fibrous plate and Roux-en-Y anastomosis of jejunum to the remaining ducts to allow for biliary drainage (Fig-1 shows pre-operative and Fig-2 shows the successful Kasai procedure).
Even with early treatment, the result of bilioenteric drainage procedures has been discouraging in the long term. Portoenterostomy done in older children has been largely unsuccessful all over the world. The poor results of Kasai prompted the search for alternative treatment and liver transplantation has emerged as a viable treatment option both as a primary procedure and after failed portoenterostomy.
The Canadian Pediatric Hepatology Research Group (CPHRG) recently discovered that 10-year native liver survival rates declined with increasing age at the time of portoenterostomy; 49% of infants who underwent surgery in Canada at 30 days of age were living with their own liver 10 years later, compared with 25% of those whose operations occurred at 31 to 90 days of age and 15% of those treated after 90 days of age.[5] Therefore, portoenterostomy should be done before there is irreversible sclerosis of the intrahepatic bile ducts. Consequently, a prompt evaluation is indicated for any infant older than 14 days with jaundice to determine if conjugated hyperbilirubinemia is present. If infectious, metabolic, endocrine disorders are unlikely and if the child has findings consistent with biliary atresia, then a surgeon who has experience doing the Kasai portoenterostomy should do exploratory laparotomy and intra-operative cholangiogram expeditiously.
Although this clinical approach leads to successful Kasai procedure in a significant number of infants however, it produces unnecessary trauma of laparotomy in many neonates who are suffering from neonatal hepatitis with a patent biliary tree in their per-operative cholangiogram. Thus a new evidence based clinical algorithm is required where neonatal hepatitis can be accurately diagnosed and differentiated from biliary atresia. Serial liver function tests do not provide any clue to diagnosis except indicating the severity of the disease. Serum values of total bilirubin, direct bilirubin, GGT, alkaline phosphatase and the alkaline phosphatase to gamma glutaryl transaminase (GGT) ratio do not reliably distinguish biliary atresia from neonatal hepatitis. Intubation and duodenal bile aspiration, though simple procedures, are relatively invasive in a newborn and a negative test (no bile aspiration) is not always specific for biliary atresia. However, bile is aspirated from the duodenum in approximately about 80% of the infants with neonatal hepatitis, but not from infants with biliary atresia.[6]
A preferably noninvasive diagnostic approach with high accuracy is desirable. Ultrasonography and 99mTc-mebrofenin scan play an important role, either in tandem or as parallel investigations, to diagnose EHBA with relatively high accuracy. Ultrasonography is a simple noninvasive first-line investigation in a neonate that can help diagnose biliary sludging, inspissated bile or gallstones and discern structural abnormalities such as choledochal cysts. A small or absent gallbladder on hepatic ultrasound suggests biliary atresia. A triangular or tubular-shaped echogenic density in the vicinity of the portal vein, on longitudinal or transverse scanning was considered a positive “triangular cord sign”. A triangular cord that was 4 mm or greater in thickness was considered a positive triangular cord sign. Lee et al (2003) and Tamazawa et al (2007) reported that the triangular cord sign, identified at the porta hepatis during ultrasound and probably representing fibrosis at the portal plate, was 73 to 100% sensitive and 98 to 100% specific for the diagnosis of biliary atresia.[7-8] Contrary to these findings, Humphrey et al (2007) reported that the “triangular cord sign” has sensitivity of 23% but with a high specificity for diagnosing biliary atresia.[9] Similar observation has also been made in a recently published large prospective study (99 infants aged less than 90 days) from India by Mittal et al. They have demonstrated the “triangular cord sign” sensitivity of 23% and specificity of 97%.[10] Thus a sensitivity as low as 23% indicates that ultrasound cannot be used to rule out biliary atresia.
Cholescintigraphy, popularly known as ‘HIDA scan’, is recoammended in all neonates with persistent jaundice beyond the second week of life or new jaundice that develops at 3 weeks after birth. Hepatospecific imino-diacetic-acid (HIDA) is a lidocaine analogue, competes with bilirubin for excretion in anionic channel of hepatocyte, and subsequently behaves like bile. Since it is labeled with technetium-99m, the radiopharmaceutical (99mTc-mebrofenin) thus delineates the biliary tree. Baseline or unstimulated HIDA scan has relatively low accuracy of about 60%. To improve the accuracy of diagnosis of neonatal hepatitis, biliary secretagogues (choleretics) are administered prior to HIDA scan and the process is called priming. The pharmacological agents employed include phenobarbital, UDCA, betamethasone, etc. An ideal patient preparation includes treatment with phenobarbital 5 mg/kg body weight/day in two equally divided doses, for 5–7 days prior to cholescintigraphy. In our own experience we have reported that adding betamethasone drops (2.5 mg/kg/day) to phenobarbital has more potent biliary stimulation and improves accuracy by another 15-20%.[11] This is based on the rationale that choleretics increase secretion of the radiotracer into bile, enabling better delineation of bile ducts and the duodenum in infants with neonatal hepatitis, but not in those with EHBA. Ursodeoxycholic acid (UDCA) administered at 20 mg/kg/day in 2-3 divided doses for 48-72h has recently being promoted as a potent stimulant of bile secretion.[12] Phenobarbital makes the child drowsy whereas UDCA has no such side effects.
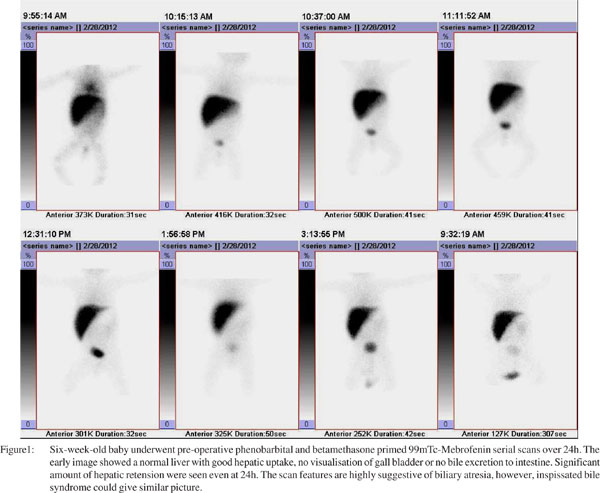
Neonates with EHBA maintain a relatively high extraction Tc-99m-mebrofenin with clear delineation of the hepatic morphology. Major abnormalities are confined to bile secretion. Despite a good extraction by the hepatocytes, there is total lack of secretion of Tc-99m-HIDA into the bile, in spite of the child being properly primed, resulting in non-visualization of the entire biliary tree. The urinary tract is an alternate channel for bile excretion, thus visualization of both kidneys with urinary bladder is very common. Delayed images at 24h do not show any evidence of bile entry into the small intestine. HIDA scan is a very good investigation to diagnose neonatal hepatitis if bile drains into the duodenum/intestine however, absence of radioactive bile in the small gut even at 24h does not offer a positive diagnosis of EHBA. HIDA scan only suggests a list of differential diagnosis namely EHBA, inspissated bile syndrome, severe cholestatic jaundice, intrahepatic biliary atresia, etc. The reported sensitivity, specificity, accuracy, positive and negative predictive value of HIDA scan is varies widely from center to center depending on the method of reparation, radiotracer used, and type and time of acquisition protocol followed. To harmonize all these protocols, the Society of Nuclear Medicine (SNM) has recently published guidelines on hepato-biliary scintigraphy.[13]
In this issue of Tropical Gastroenterology, Shah et al have studied 46 neonates, who presented with conjugated hyperbilirubenemia.[14] They have used 99mTc-mebrofenin scan to diagnose various causes of jaundice and validated the findings against per operative cholangiography. The title, “Utility of Tc99m-mebrofenin hepato-biliary scintigraphy (HIDA scan) for the diagnosis of biliary atresia” is very apt. A modest attempt to justify the regular use of HIDA scans has been made for the work-up of neonatal jaundice. The accuracy of diagnosis by HIDA scan (proper classification) reported by the authors is 33/46 (71.7%) which is an average value reported by several investigators. I am really intrigued to see a large number of neonates (6/28 i.e. 21.4%) had bile secretion into the small gut but were subsequently diagnosed as biliary atresia.
The conventional aphorism of biliary atresia diagnosis is the absence of radioactive bile in small gut. If bile is detected at any point of time during the study period (up to 24h), it rules out the diagnosis of biliary atresia. Contrary to belief, 39% proven neonatal hepatitis cases demonstrated no gut secretion, which is far greater than expected. For such a high proportion of non-secretory cases one could argue that the neonates were perhaps not properly primed. However, faulty preparation method is completely ruled out and moreover, both choleretics phenobarbital and UDCA in adequate doses were concurrently administered to each neonate in this series. It is a well established fact that neonatal hepatitis far exceeds biliary atresia in any published series including several from India. It is also very difficult to appreciate why Shah et al had very large number of EHBA (28/46 i.e. 61%) in their series; could it be a referral bias or children with high degree of suspicion of EHBA were investigated for per-operative cholangiogram at a tertiary care centre for Kasai procedure?
 In spite of poor sensitivity of abdominal sonography, it is recommended in all neonates with conjugated hyperbilirubinemia to diagnose biliary atresia (triangular cord sign and absence of gall bladder). 99mTc-mebrofenin (HIDA) scans are recommended to “rule in” neonatal hepatitis. The combination of both shall prevent many unnecessary peroperative cholangiograms.
References
In spite of poor sensitivity of abdominal sonography, it is recommended in all neonates with conjugated hyperbilirubinemia to diagnose biliary atresia (triangular cord sign and absence of gall bladder). 99mTc-mebrofenin (HIDA) scans are recommended to “rule in” neonatal hepatitis. The combination of both shall prevent many unnecessary peroperative cholangiograms.
References
- Craig JM, Landing BH. Form of hepatitis in neonatal period simulating biliary atresia. AMA Arch Pathol. 1952;54:321–33.
- Cocjin J, Rosenthal P, Buslon V, Luk L Jr, Barajas L, Geller SA, et al. Bile ductule formation in fetal, neonatal, and infant livers compared with extrahepatic biliary atresia. Hepatology. 1996;24:568–74.
- Practice parameter: management of hyperbilirunemia in the healthy term newborn. American Academy of Pediatrics. Provisional Committee for Quality Improvement and Subcommittee on Hyperbilirunemia. Pediatrics. 1994;94:558–65.
- Arora NK, Kohli R, Gupta DK, Bal CS, Gupta AK, Gupta SD. Hepatic technetium-99m-mebrofenin iminodiacetate scans and serum gamma-glutamyl transpeptidase levels interpreted in series to differentiate between extrahepatic biliary atresia and neonatal hepatitis. Acta Pediatr. 2001;90:975–81.
- Schreiber RA, Barker CC, Roberts EA, Martin SR, Alvarez F, Smith L, et al. Biliary atresia: the Canadian experience. J Pediatr. 2007;151:659–65, 665.e1.
- Greene HL , Helinek GL , Moran R , O’Neill J. A diagnostic approach to prolonged obstructive jaundice by 24-hour collection of duodenal fluid. J Pediatr. 1979;95:412–4.
- Lee HJ, Lee SM, Park WH, Choi SO. Objective criteria of triangular cord sign in biliary atresia on US scans. Radiology. 2003;229:395–400.
- Takamizawa S, Zaima A, Muraji T, Kanegawa K, Akasaka Y, Satoh S, et al. Can biliary atresia be diagnosed by ultrasonography alone? J Pediatr Surg. 2007;42:2093–6.
- Humphrey TM, Stringer MD. Biliary atresia: US diagnosis. Radiology. 2007;244:845–51.
- Mittal V, Saxena AK, Sodhi KS, Thapa BR, Rao KL, Das A, et al. Role of abdominal sonography in the preoperative diagnosis of extrahepatic biliary atresia in infants younger than 90 days. AJR Am J Roentgenol. 2011;196:W438–45.
- Gupta DK, Charles AR, Srinivas M, Dave S, Bal CS. Betamethasone plus phenobarbitone prior to hepatobiliary scintigraphy increases diagnostic accuracy in infants with jaundice. Indian J Pediatr. 2001;68:1039–41.
- Poddar U, Bhattacharya A, Thapa BR, Mittal BR, Singh K. Ursodeoxycholic acid-augmented hepatobiliary scintigraphy in the evaluation of neonatal jaundice. J Nucl Med. 2004;45:1488–92.
- Tulchinsky M, Ciak BW, Delbeke D, Hilson A, Holes-Lewis KA, Stabin MG, et al. SNM practice guideline for hepatobiliary scintigraphy 4.0. J Nucl Med Technol. 2010;38:210–8.
- Shah I, Bhatnagar S, Rangarajan V, Patankar N. Utility of Tc99mmebrofenin hepato-biliary scintigraphy (hida scan) for the diagnosis of biliary atresia. Trop Gastroenterol. 2012;33:62–4.