|
|
|
|
 |
|
| |
 |
|
|
Quarterly Reviews |
|
|
|
|
|
Keywords :
hepatic fibrosis, chronic liver disease, antifibrotic therapy |
|
|
K. Das, Abhijit Chowdhury
Division of Hepatology
School of Digestive and Liver Diseases
Institute of Post Graduate Medical Education & Research,
Kolkata.
Corresponding Author:
Dr. Abhijit Chowdhury
Email: achowdhury2002@yahoo.co.in
DOI:
http://dx.doi.org/
Abstract
Hepatic fibrosis, a reparative response to different types of liver injury, has emerged as the primary determinant of outcome in advancing chronic liver disease, including cirrhosis. Once considered irreversible, today there is enough clinical as well as laboratory data available for us to be optimistic and expect regression of liver fibrosis in clinical situations, with resultant improvement in outcome. The primary premise of this approach to modify liver fibrosis has been its success in treating the basic pathology underlying persistent liver inflammation and injury, often with the reversal of cirrhosis. However, more focused anti-fibrotics altering the dynamics of collagen deposition and resorption are undergoing evaluation and will be available shortly. In this changing scenario, there is a need for precise, easy to use endpoints of success / failure of anti-fibrotic therapy. A future scenario may be envisaged as one of a more positive, aggressive approach to treatment of chronic liver disease- treating the cause as well as using anti-fibrotics. This “hit the enemy and repair the hut” approach ushers in a new era from the hitherto barren pessimism in the treatment of chronic liver disease.
|
48uep6bbphidvals|224 48uep6bbphidcol2|ID 48uep6bbph|2000F98CTab_Articles|Fulltext There have been spectacular advances in our understanding of the pathology of liver fibrosis over a short period of time. Our success in treating hepatitis B and C, the major causes of liver fibrosis has created a need for therapy directed at the basic biology of fibrosis per se. Our present dilemma may be captured in the following sentence: “The nemy is being taken care of, but can we reconstruct the house damaged and destroyed?” Scientific enthusiasm is often idealistic and driven by passion and emotions; clinical realities are harsher. This is becoming increasingly relevant in the era of biotechnology where a large number of techniques and molecules are constantly being developed, each with a different hue and a different hope. In a disease, as morbid as cirrhosis, any new development is received with cautious optimism. The discussion of the developments that follow is thus based on this tentative optimism.
Reversibility of liver fibrosis: Genesis and evidence (Figure 1)
The sequence of scientific progress in medical science usually is as follows: clinical observation and anecdotes giving birth to notions and ideas, basic scientist takes cue, hypothesises, experiments, infers, exchanges views with clinical scientist, goes back to the laboratory, modifies and improves scientific steps, applies initially with skepticism and thereafter with vigour, rationalises and hands over concept to clinical scientist for application and translation. Hepatic fibrosis research has also long obeyed these sound principles. Liver fibrosis and cirrhosis have long been considered a prelude to certain death. An astute observer, Ruy Perez Tamayo, lamented in 1965 “Perhaps our concepts about connective tissue are more resistant to change, more permanent and irreversible than collagen fibers themselves. At least the later can be resolved.” After Perez Tamayo’s hypothesis1 of ‘dynamic disturbance in morphostasis’ and potential reversibility of cirrhosis, Wanless[2] documented reversibility of cirrhosis in a chronic hepatitis B patient. Notions in the concept of fibrosis reversibility became convictions after Poynard reported reversal of fibrosis in chronic hepatitis C (CHC) patients.[3] Later on reversal of fibrosis was reported in 49% of CHC patients with baseline cirrhosis who were sustained responders, treated for 48 weeks and were younger.[4] A number of studies have subsequently shown regression of fibrosis where necroinflammatory activity is reduced by the available therapies e.g. in chronic hepatitis B and C, autoimmune hepatitis, Wilson’s disease and alcoholic liver disease.[2,4,5]
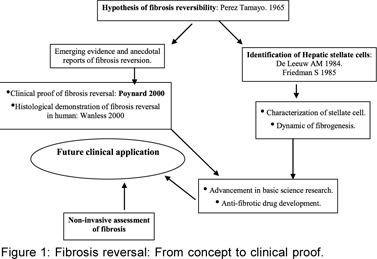
These clinical studies around the same period were supported by animal experiments and laboratory experiments. Liver fibrosis was delineated as a dynamic process even in advanced cirrhosis. An altered micro environmental milieu sets as well as sustains a cascade of signals, often with positive and negative feedback effects, involving the stellate cell in the centre of action. The stellate cell, in turn, receives input and influences from the hepatocytes, inflammatory cells and migrating cells and the constituents of the scalloping called the extracellular matrix, which it produces. There is balanced production and resolution of fibrous tissue, which process goes on for a while and the net of these two counterbalancing acts, i.e fibrogenesis and fibrolysis, determines the biology of progression or regression of the fibrosis. Scott Friedman and John P Iredale had provided the cellular and molecular evidence for reversibility of fibrosis. It also has become evident that it is possible to modulate the micro environmental milieu in the liver, by influencing the extracellular signals impinging on the stellate cell and also by changing the’ phenotype – from an aggressively active one to one committed to death and demise by apoptosis. This opened up the new avenue – the era of anti fibrotics – that are expanding everyday.
Meanwhile, the mechanism of fibrosis reversal from a histologic point of view has been described in intricate detail by Wanless.[2] The regression parameter termed ‘Hepatic repair complex’ was logically derived from histologic data. Micronodular cirrhosis, macronodular cirrhosis and incomplete septal cirrhosis were part of a continuum. Four patterns: sinusoidal fibrosis, septal fibrosis, fibrous adhesion and large areas of extinction were described. These may form a continuum. A picture of large areas of extinction consisting of proliferating ductules, hepatocytes surrounded by collagenous matrix and vascular channels was found to be least amenable to reversal. In their study, livers with incomplete septal cirrhosis (ISC) showed features of ‘hepatic repair complex’ including ‘venoportal adhesions’ and aberrant veins which were not expected in early stages of ‘cirrhosis.’ So, logically ISC appeared to be a regressed form of cirrhosis.[2]
This paradigm shift in the approach to hepatic fibrosis warrants a system of appraisal that is accurate, hassle free, reproducible and able to keep pace with the needs of therapy over a longitudinal time frame. Liver biopsy, the traditional gold standard, has therefore been supplemented with a number of new assessment modalities, called “noninvasives”, that has ushered in a new era of fibrosis assessment. Imaging methods such as ultrasound elastography and MR elastography as well as methodologies like glycomics, proteomics are being increasingly evaluated in the search for a “biomarker” of liver fibrosis that can precisely guide therapy, including monitoring of therapy effectiveness.
Factors influencing fibrosis progression and regression
The fibrosis – cirrhosis controversy
The term ‘cirrhosis’ is often loosely used in medical literature. Research in the early nineties visualised cirrhosis as ‘sclerosed liver’ and the term liver fibrosis came in to vogue.[6,7] Functional abnormalities of chronic liver disease could not be explained by the morphological concept of fibrosis only. A consensus meeting held in 1956 determined that ‘fibrosis should not be used synonymously for cirrhosis’ and recognised that ‘cirrhosis’ implied much more than advanced fibrosis and included vascular reorganisation, neovascularisation, parenchymal loss and regeneration.[7,8]Presently ‘fibrosis’ is considered a component of ‘cirrhosis’ Fibrosis then is potentially reversible but the very minimal or slow regression potential of ‘large extinct areas’, portocentral vascular bridging and neovascularisation seen in cirrhosis question its reversibility, particularly so because of the reduced life span of patients in this particular group.
Biological basis of selecting therapeutic targets (Figure 2)
Hepatic fibrosis is the net acquisition of abnormal collagen in the liver, in abnormal proportions and unusual locations impeding its functioning and culminating into cirrhosis. Regression of fibrosis means reduction of collagen content, often with changes in collagen tissue composition with potential for improvement in cell and organ functioning and delay/discontinuance in the development of clinical events. Fibrosis regression may be histological regression alone or regression with functional improvement as manifested by reduced hepatic vein pressure gradient, improving liver synthetic function, reduced complications and prolonged survival. Pathophysiological determinants for reversal are apoptosis of the activated stellate cell, degradation of the neo-matrix rich in type I collagen with grossly increased matrix metalloproteinase activity, removal of the offending agent and regeneration of the liver parenchyma.[9,10]

Extracellular matrix is the scaffolding for collagen deposition and resorption
Extracellular matrix (ECM) consists of collagen, glycoprotein, proteoglycans, and glycosaminoglycans. It provides a complex for different growth factors, cytokines, matrix metalloproteinase, tissue inhibitors of metalloproteinase and tissue transglutaminase. The ECM is not inert. It modulates stellate cell behaviour through the transmembrane receptor, integrin.[11] Many integrin ligands contain an Arg-Gly-Asp (RGD) tripeptide sequence. Integrins have been identified in hepatic stellate cells and myofibroblasts and this has led to experimental attempts to develop RGD peptide analogues to inhibit fibrosis.[12]
Stellate cells play a dual role and are crucial in fibrogenesis as well as fibrolysis
Stellate cells are activated from a quiescent state to a myofibroblast like cell that is central in fibrogenesis. Different cytokines and inflammatory mediators that maintain a profibrogenic environment, help maintain an activated stellate cell phenotype. Studies in rat stellate cells show that in quiescent form they express mRNA for procollagen I & III at very low levels, are less proliferative and are less responsive to cytokines and growth factors. Studies both in animal models and human fibrogenesis show a positive correlation between the extent of fibrosis and the accumulation of activated hepatic stellate cells in the area of injury.[13] Initiation of activation is by paracrine stimuli from injured hepatocytes, Kupffer cells, platelets and infiltrating inflammatory cells. Once activated, stellate cells maintain their phenotype by upregulating growth factor and cytokine receptors and by positive interaction with the extracellular matrix. Mitogens like plateletderived growth factor (PDGF), fibroblast growth factor, thrombin, angiotensinogen and others promote proliferation over apoptosis.[13] Loss of active phenotype of hepatic stellate cell is a major determinant for fibrosis regression. This can be achieved by either inhibiting its proliferation or by inducing apoptosis.[14,15] After CC[l4] treatment, a proportion of myofibroblasts derived from hepatic stellate cells seems to dedifferentiate while in the bile duct ligation model, myofibroblasts derived from portal fibroblasts disappear by apoptosis, underlining the relevance of this model to evaluate the mechanisms involved in fibrotic liver remodelling.[15] Antagonists for PDGF receptor and different apoptosis inducing agents are being tried in animal models towards producing fibrosis reversal.
MMP’S, TIMPs, apoptotic protein regulators and other signals modulate outcome (Figure 3)
Matrix metalloproteinase (MMPs) and tissue inhibitor of metalloproteinase (TIMPs) in unison produce ‘pathological matrix degradation’ and inhibition of ‘restorative matrix degradation’ thereby allowing progressive fibrogenesis.[16] Normal low density lattice like extracellular matrix is replaced by high density scar tissue resulting in nodularity, vascularisation and cellular septae leading to deterioration of hepatic function. Collagen cross-linking by the action of
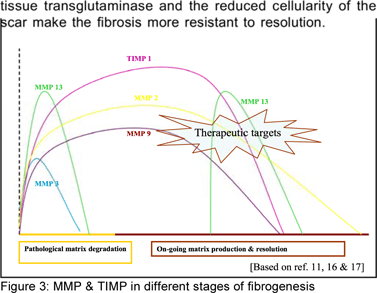
Every step is being explored to develop anti-fibrotic agents. A major area of concern is modulation of the stellate cell behaviour and the cytokine milieu. A systematic review of literature on expression and modulation of MMPs and TIMPs showed that complete reversibility of fibrosis is possible in mouse and rat models[16] and that this modulation may interrupt the vicious cycle of liver fibrogenesis and enhance fibrolysis.[17]
What do animal models tell us?
Animal models of experimental fibrogenesis show that complete reversibility of fibrosis is possible. In health, constant interaction among MMPs and TIMPs maintains the integrity of the tissue matrix. Imbalance resulting from chronic liver injury results in differential expression of MMPs and TIMPs, thus leading to fibrogenesis. Promising anti-fibrotic approaches modulating those enzymes showed improvement in soluble and immunohistochemical fibrosis markers. Reduction of TIMP-1, MMP-2 activity and increase in collagenase, MMP-3 & 13 activity correlated with reduction in collagen I and III, fibronectin and a-smooth muscle actin staining in different models.[17] The picture in the human subject is expected to be more complicated.
Clinical fibrosis – determinants of behaviour
Quality of the fibrotic tissue: Older collagens with cross-linking because of tissue transglutaminase activity are relatively resistant to resolution. Hypocellularity of the scar tissue also deprives the ECM of available metalloproteinase and other enzymes responsible for restorative matrix degradation.
Aetiology and histology
An important clinical question is whether the progression of fibrosis depends on the aetiology of liver injury or not. The pattern and behaviour of fibrosis has been best studied in HCV infection. In order to achieve objectivity in histological assessment of progression/regression of fibrosis, European workers have evolved the Metavir score, while other systems (Ishak, Scheur’s) may also be used interchangeably. Each of these scales provides a numerical value to biological events and thereby helps studying changes over a particular time period. Fibrosis progression is not linear and the rate of progression is not uniform. However, there can still be rapid, intermediate and slow fibrosers,[18,19] based on genetic and concurrent environmental factors. Most importantly, age of acquisition of infection, male sex, co-infection with HBV/HIV, alcohol use, fat mass, and iron over load are factors adversely influencing outcome of HCV fibrosis. While generalizations cannot be made and firm conclusions cannot be drawn on the influence of aetiology and concurrent factors in disease progression, this information may help in therapy stratification, along with other factors.
Host genetic polymorphisms[20,21]
Many different single nucleotide polymorphisms, particularly those in the genes encoding for proinflammatory cytokines, immunoregulatory proteins, metabolic enzymes and fibrogenicfactors, may play an important role in fibrosis[ 20] However, the strength
Irreversible fibrosis:
Perez-Tamayo remarked that collagen cross-linking made the scar hard to resolve.1 Wanless demonstrated that large areas of extinction hinder fibrosis reversibility. Incomplete septae and macronodular cirrhosis may actually comprise a regressed form of advanced fibrosis.[2] Experimentation using the CCl4 treated rat model showed that continued toxin exposure delayed fibrosis reversal due to the presence of thick cross-linked collagen with high levels of tissue transglutaminase and TIMP-1.[22]
Regression of fibrosis: clinical relevance (Figure 4)
Regression of fibrosis is demonstrable histologically in compensated liver disease. But regression to what extent is clinically meaningful is not yet clear. Regression, to be called clinically significant, must have some functional correlate, like reduction in portal pressure and improvement in liver function test results. More important would be a reflection of histological and functional regression in ‘clinical regression’. In clinical regression we should expect reduced number of clinical events like variceal bleeding, hepatic encephalopathy and other decompensating features in the short term and a reduction in the risk of HCC development and improved overall survival in the long term.
The therapeutic response in different aetiologies improves clinical out come and survival. This has been most dramatically demonstrated in decompensated chronic hepatitis B. Lamivudine therapy has been shown to improve child status, diminish/ delay transplant needs and improve survival. However, the biological basis of this is yet to be explained. Moreover, it needs to be determined whether these changes are due to the impact of therapy on fibrosis per se or some other disease determinant. Advanced cirrhosis of liver is a multi-system disease with derangements in metabolic, immunological and functional milieu in many organ systems. Each of these is interconnected. The pathophysiological basis of improvement with therapy in the hepatic and extra hepatic sites needs to be clearer for his observation to be practically useful. We need to define our target for fibrosis reversibility and to what extent reversibility is reflected in clinical outcome, and we need to find ways to measure this.
Assessment of fibrosis: Is there a better way?
Percutaneous liver biopsy is still the gold standard for fibrosis assessment. In the face of exuberant development of noninvasive modalities we need to choose the best option for clinical practice. The Ishak, Metavir and Scheuer scores are the most widely used scoring systems. They were developed before the feasibility of fibrosis reversal was so widely acknowledged. Sampling error in histology may be in the range of 33% to 50%. One study using laparoscopic liver biopsy showed discrepancy of fibrosis by at least one stage between the two lobes.[23] Sampling errors may also hamper the assessment of non-alcoholic fatty liver disease.[24] A biopsy sample is about 1/50,000th of the liver volume and thus sampling error is inevitable. Conventional histological scores were not validated in assessment of fibrosis regression. These deficiencies necessitated the development of noninvasive markers.
Non-invasive markers for fibrosis:
Surrogate markers singly or in combination e.g. APRI (aspartate aminotransferase to platelet ratio index), Forn’s index and Fibrotest are indirect indications of fibrosis whereas direct biomarkers e.g. hyaluronic acid, collagen IV, procollagen I carboxy terminal peptide, MMP and TIMP assess fibrosis biology directly. A combination of direct and indirect markers has been used in FibrospectII, European liver fibrosis and Hepascore. Biomarkers may reflect the changes in fibrogenic activity before they are reflected in absolute matrix content in scar tissue. Clinical management demands correlation of biomarkers with the complications of progressive fibrosis and not just a simple quantitative assessment of fibrous tissue.
A review of the available literature shows that APRI was able to exclude cirrhosis in 85% of the patients.[25] Another one reported 94% accuracy in detecting fibrosis above F2.[26] On the contrary, APRI was less sensitive in a French cohort.[27] Forn’s index which includes age, gamma GGT, cholesterol and platelet count was found to estimate F2 to F4 stages in less than 40% of patients. Retrospective study in chronic hepatitis B patients revealed a Fibrotest score less than 0.2 and more than 0.8 had a negative predictive value of 92% and a positive predictive value of 92% for diagnosis of cirrhosis and hence 46% of liver biopsies can be avoided on the basis of the Fibrotest score.[28] Another study in an HCV cohort reported 95% accuracy in diagnosing cirrhosis; and determined that 60% of the liver biopsies may have been prevented if APRI, Fibrotest and Forn’s index had been combined in an algorithm.[26]
Interestingly, Fibrotest score <0.75 excluded the presence of large oesophageal varices with a negative predictive value of 100%.[29] A multicentric study with chronic hepatitis C patients validated the use of a combination of hyaluronic acid, TIMP-1 and a-2 macroglobulin and showed an accuracy of 75% in differentiating between F0-F1 and F2-F4 stages. But interestingly 14% of the patients would have been misclassified as F2-F4 fibrosis; conversely, 11% would have been misclassified as minimal fibrosis cases.[30]
Too many markers, too many combinations mean none of them are efficient enough to be used clinically on a regular basis. It is obvious that minimal/no fibrosis can be confidently differentiated from cirrhosis by non-invasive tests. Even with substantial improvement in their development and accuracy they cannot replace liver biopsy at present. To treat fibrosis clinically, we need to define every stage so as to screen for its sequelae. However, it is very difficult to compare these continuous values of non-invasive tests with descriptive Metavir/Ishak stages. Thus poor correlation between them is quite expected. Moreover a majority of the studies are not aimed at defining intermediate stages. Extrapolating the data in different situations will not provide a true picture.
Role of Radiology
A promising way to estimate liver fibrosis is to measure liver stiffness by transient elastography. Fibroscan uses low frequency vibration and fibrosis is measured in kilopascals. It measures liver stiffness within a volume of 1cm x 2cm approximately, which is comparatively larger than an average liver biopsy.
Meta-analysis of 9 studies showed sensitivity and specificity of 87% and 91% respectively to detect cirrhosis by transient elastography.[31] In a population of 1007 patients with compensated cirrhosis of different aetiologies a liver stiffness cut off value of 14.6 kPa had specificity of 95%, positive predictive value of 74% and negative predictive value of 96% with an accuracy of 92% for the diagnosis of cirrhosis.[32]
It appears that exclusion of cirrhosis was more accurate than its diagnosis. False positive results were found in cases of extensive fibrosis and false negative in cases with macronodular cirrhosis. Important information that derived from the study was that specific cut-off values can be defined for specific aetiologies.[32] Differences in cut-off values for diagnosis of cirrhosis varied among aetiologies as reported by other studies.[33] Estimation at more than one time point is required to appreciate the exact stiffness value because acute exacerbation of hepatitis activity may increase the stiffness by 1.3 to 3 times[34] In another recent study Fibroscan misclassified 15 non-cirrhotic patients as cirrhosis cases.[35] In the ‘proof of principle’ study by Yeh et al, it was suggested that “biological tissue is a composite material and it is difficult to separate the influence of each component of the tissue on the total of modulus estimates”.[36] At the moment we cannot measure the portal pressure/HVPG in terms of liver stiffness as different dynamic mechanical and biochemical factors, and splanchnic as well as collateral circulations are involved. However, a liver stiffness value of >13.6 kPa correlates with clinically significant portal hypertension (HVPG > 10 mm Hg) and this correlation is more significant at HVPG < 10-12 mm Hg.[37] A stiffness value of < 19 kPa was able to exclude the presence of large oesophageal varices with a negative predictive value of 93% [38] but failed to assess the variceal size. Foucher reported correlation between liver stiffness and the presence of oesophageal varices and also with history of bleeding from varices.[39] What we can derive from the literature is that liver stiffness in the cirrhotic range does not always mean advanced fibrosis. Further studies are needed to see the effect of steatosis and acute inflammatory activity on stiffness measurement.
Another new but simple technique to measure hepatic vein transit time (HVTT) using micro-bubble ultrasound is reported to correlate with increasing liver fibrosis.[40]
Perfusion imaging with CT and MRI has the potential to detect hemodynamic changes because of perisinusoidal fibrosis and can detect early stages of fibrosis before cirrhosis sets in.[41] In comparison to ultrasound based elastography magnetic resonance elastography can scan the whole liver avoiding sampling bias. But this modality cannot define intermediate stages of fibrosis, is not costefficient and is therefore unlikely to be acceptable to patients.[42]
Proteomics and glycomics in hepatic fibrosis
Proteomics and microarray technologies are in too primitive a stage to be applied to clinical practice. Preliminary studies using DNA-sequencer based total serum protein glycomics could differentiate between compensated chronic liver disease and non-cirrhotic chronic liver disease by identifying modification of the five peaks of the N-glycome profile.[43] But this was not useful for identifying lesser stages of fibrosis. Proteomics and glycomics technologies need further development to be at par with other non-invasive markers at least.
So, till date we can at least differentiate between minimal or no fibrosis and advanced fibrosis by using oninvasivemarkers or imaging techniques and thereby guide our therapeutic decision. More refined procedures and tests are required to allow us to pick up intermediate stages. With so many non-invasive modalities the approach to clinical management of liver fibrosis is likely to change in the near future. The best approach will be to use a combination of the tests previously mentioned; this combination will be applied clinically, be stable and reproducible, and will reflect the dynamic nature of fibrosis, and be efficient in detecting the intermediate stages.
Current status of anti-fibrotic therapy
Treatment of the specific cause
Regression of fibrosis is documented in chronic liver disease wherever aetiology specific therapy is possible to halt the necro-inflammatory activity as in HBV, HCV, autoimmune hepatitis, Wilson’s disease and hemochromatosis.
Targeted anti-fibrotic therapy (Table 1)
Extra-cellular profibrogenic factors like MMPs, TIMP and TGF- a and pro-proliferative factors like PDGF and integrin are the most attractive targets. Therapeutic intervention to change hepatic stellate cell behaviour is also a promising area. Proinflammatory intracellular signalling pathways are being explored to develop anti-fibrotic drugs.
Modulation of profibrogenic extracellular regulators
TIMP-1 neutralising antibody is found to reduce collagen synthesis in the CCl[4] treated rat model.[44] Hepatic stellate cells cultured in plastic media were protected from induced apoptosis by exogenous TIMP-1, whereas TIMP-1 neutralising antibody induced apoptosis of those stellate cells.[45] Removal of collagen tissue by up-regulating the matrix metalloproteinase is another way of treating fibrosis. Adenoviral transfection of MMP-1 and 8 in animal models attenuated liver fibrosis.[46,47] Angiotensin II acting via the AT 1 receptor induces stellate cell contraction and proliferation, increases collagen I synthesis and also induces synthesis Tropical Gastroenterology 2008.29;2:76– 83 of profibrogenic TGF-a. Candesartan, an AT1 receptor blocker and perindopril, an ACE inhibitor were observed to attenuate liver fibrosis in rat model.[48] But a small trial with losartan showed reduction in the area of fibrosis but no improvement in the fibrosis score in histology.[49] However, angiotensin receptor blockers and ACE inhibitors are well tried clinically in cardiovascular disease and thus have potential for rapid clinical development in the field of liver fibrosis.
Modulation of stellate cell behaviour: Activated stellate cells express aVß3 integrin to avail the survival signals from the extracellular matrix. Antagonism of this integrin induces apoptosis and reduced proliferation of the stellate cells.[50] Different other interventions involving cytokines, growth factors and extracellular matrix are designed to affect stellate cell functioning. Antagonism of profibrogenic TGF-a by soluble type II receptor antagonist reduces fibrosis in bile duct ligated rats.[51]
Modulation of intracellular signalling pathways
NF-?ß, a dimeric nuclear transcription factor helps to maintain activated phenotype of hepatic stellate cells and makes them resistant to apoptosis. Anti-apoptotic activity of TNF-a and TGF-ß is associated with increased activity of NF-?ß. Treatment of cultured myofibroblasts with sulfasalazine induced apoptosis of myofibroblasts in a dose dependant manner.[52] Use of sulfasalazine is promising but needs careful trial in chronic liver disease for clinical application. Nuclear receptors, PPAR-? in particular, are found to be highly expressed in quiescent hepatic stellate cells. Ligands of these receptors inhibit transdifferentiation into the activated phenotype.[53] PPAR-? agonist pioglitazone is already being used as an insulin sensitiser. So, this group of agents raised much clinical interest.
Herbal agents
Different herbal agents with presumed anti-viral, anti-inflammatory and anti-fibrotic agents have been used particularly in the Far East countries.[54] Sporadic data showed clinical efficacy. Inchin-ko-to (TJ-135) and Sho-saiko-to have been shown to improve fibrosis in animal models.[55,56] Herbs of Salvia genus (Salvia miltiorrhiza monomer IH764-3) has been used in China as anti-fibrotic.[57] Because of concern of potential hepatotoxicity herbal agents were not ried in other countries.
Anti-oxidants and cytoprotectives
Alfa tocopherol, S-adenosylmethionine, and propylthiouracil have been used in different trials where fibrosis assessment was not included as a specific outcome. Lack of hard evidence of their efficacy on fibrogenesis prevents their clinical use.[58,59,60,61]
Preclinical studies promote the concept of potential reversibility of fibrosis by interfering with the different steps of fibrogenesis. Before human application, several issues like adverse events related to the drug delivery system, metabolic effects in diseased liver and extra-hepatic unwanted effects require further research.
Future directions
Once a decision has been taken to initiate treatment of a patient with significant fibrosis, a clinician will need to be clear on several issues. Is the choice of treatment effective, simple, cheap and with predictable outcome? How will the treatment results be monitored? How long should the treatment be continued? How best to follow up? Each of these is a vexing issue – with incomplete answers at present. The anti-fibrotics are in phase II / III and more data are needed. The best treatment for fibrosis available today comprises treatment directed at the aetiology e.g. antivirals, steroids, weight reduction and alcohol abstinence. While baseline and endpoint assessments by liver biopsy provide useful information, a significant grey zone of fibrosis evolution in between needs to be assessed precisely. The serum markers, fibroscan, glycomics and proteomics have raised hope but are yet to prove their utility in discriminating the subtle changes in fibrosis behaviour – Metavir 2 /3. Moreover, it appears that each of these will have their own place in assessment and monitoring, as a complimentary / ancillary aid to histology. Histology still remains the trusted guide for baseline as well as end point assessment. Besides, before we go on to use fibrosis targeted therapies, we need to answer: what is the ideal agent? Is it a single agent or do we use a combination therapy? Moreover, we have to decide whether to treat the cause and fibrosis one at a time or to treat both together. Before using in clinical practice, ‘accurate end points’ for the trials involving anti-fibrotic agents need to be defined. Clinical end points like reduction in the risk of HCC development, reduction in portal pressure and improvement in the overall survival are required in trials to assess the effect of fibrosis regression on clinical grounds.
Till more data are available, it is best to aim at treating the Das et al: Hepatic fibrosis 81 cause, in the optimism that anti-fibrotic agents will very soon be available for clinical use. We can learn from the atherosclerosis story. Even the greatest visionaries in lipid and platelet research could not predict and anticipate the rapid translation of basic research on the interaction of lipids and platelets in coronary artery disease, to its clinical application. Today, we have effective therapy targeting dissolution of the atherosclerotic plaque. Techniques such as laser capture micro- dissection can now select out cellular and sub-cellular organelles as a research tools. Advances in imaging, drug development, drug delivery systems and targeted delivery in the liver tissue are now beyond fiction. It is probably a couple of years before we get the antifibrotic scissor, removing debris from the cirrhotic liver, to revive the hepatocytes. We will have balanced regenerators to push them on to the cell cycle and we may then have agents modifying the vascular biology.
References
1. Perez-Tamayo R. Some aspect of connective tissue of the liver. Progress in liver disease 1965.Grune & Stratton. Vol II; 192–210.
2. Wanless IR, Nakashima E, Sherman M. Regression of human cirrhosis: morphologic features and the genesis of incomplete septal cirrhosis. Arch Pathol Lab Med. 2000;124:1599–607.
3. Poynard T, McHutchison J, Davis GL, Esteban-Mur R, Goodman Z, Bedossa P, et al. Impact of interferon alfa-2b and Ribavirin on progression of liver fibrosis in patients with chronic hepatitis C. Hepatology. 2000;32:1131–7.
4. Poynard T, McHutchison J, Manns M, Trepo C, Lindsay K, Goodman Z, for the PEG-FIBROSIS Project Group, et al. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology. 2002;122:1303–13.
5. Dufour JF, DeLellis R, Kaplan MM. Reversibility of hepatic fibrosis in autoimmune hepatitis. Ann Int Med. 1997;127:981–5.
6. Mallory FB. Cirrhosis of the liver. Five different types of lesions from which it may arise. Bull John Hopkins Hosp. 1911;22:69–75.
7. Fifth Pan American Congress of Gastroenterology, Report of the Board for Classification and Nomenclature of Cirrhosis of the Liver. Gastroenterology. 1956;31:213–219. La Habana, Cuba.
8. Sherman IA, Pappas SC, Fisher MM. Hepatic micro vascular changes associated with the development of liver fibrosis and cirrhosis. Am J Physiol. 1990;258:H460–H465.
9. Iredale JP. Hepatic stellate cell behavior during resolution of liver injury. Semin Liver Dis. 2001;21:427–436.
10. Guo J, Friedman SL. Hepatic Fibrogenesis. Semin Liver Dis. 2007;27:413–26.
11. Schuppan D, Ruehl M, Somasundaram R, Hahn EG. Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis. 2001 Aug;21(3):351–72.
12. Iwamoto H, Sakai H, Tada S, Nakamuta M, Nawata H. Induction of apoptosis in rat hepatic stellate cells by disruption of integrinmediated cell adhesion. J Lab Clin Med. 1999;134:83–9.
13. Ramón Bataller, David A. Brenner. Hepatic Stellate Cells as a Target for the Treatment of Liver Fibrosis. Semin Liver Dis. 2001. Vol 21; No 3: 437–451.
14. Iredale JP. Hepatic stellate cell behavior during resolution of liver injury. Semin Liver Dis. 2001;21:427–436.
15. Guyot C, Combe C, Balabaud C, Bioulac-Sage P, Desmoulière A. Fibrogenic cell fate during fibrotic tissue remodeling observed in rat and human cultured liver slices. J Hepatol. 2007;46:142–150.
16. Friedman SL. Liver fibrosis – from bench to bedside. J Hepatol. 2003;38 Suppl 1:S38–53.
17. Hemmann S, Graf J, Roderfeld M, Roeb E. Expression of MMPs and TIMPs in liver fibrosis – a systematic review with special emphasis on anti-fibrotic strategies. J Hepatol. 2007;46:955–75.
18. Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet. 1997;349:825–32.
19. Fattovich G, Giustina G, Degos F, Tremolada F, Diodati G, Almasio P, et al. Morbidity and mortality in compensated cirrhosis type C: a retrospective follow-up study of 384 patients. Gastroenterology. 1997;112:463–72.
20. Bataller R, North KE, Brenner DA. Genetic Polymorphisms and the Progression of Liver Fibrosis: A Critical Appraisal. Hepatology 2003;37:493–503.
21. Christoph H.O¨ sterreicher, Felix Stickel, and David A. Brenner. Genomics of Liver Fibrosis and Cirrhosis. Semin Liver Dis 2007. Vol 27; No 1: 28–43.
22. Issa R, Zhou X, Constandinou CM, Fallowfield J, Millward-Sadler H, Gaca MD, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology 2004;126:1795–1808.
23. Regev A, Berho M, Jeffers LJ, Milikowski C, Molina EG, Pyrsopoulos NT, et al. Sampling error and intraobserver variation in liver biopsy in patients with chronic HCV infection. Am J Gastroenterol. 2002;97:2614–8.
24. Ratziu V, Charlotte F, Heurtier A, Gombert S, Giral P, Bruckert E, et al. Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology. 2005;128:1898–906.
25. Lackner C, Struber G, Liegl B, Leibl S, Ofner P, Bankuti C, et al. Comparison and validation of simple noninvasive tests for prediction of fibrosis in chronic hepatitis C. Hepatology. 2005;41:1376–82.
26. Sebastiani G, Vario A, Guido M, Noventa F, Plebani M, Pistis R, et al. Stepwise combination algorithm of non invasive markers to diagnose significant fibrosis in chronic hepatitis C. J Hepatol. 2006;44:686–93.
27. Le Calvez S, Thabut D, Messous D, Munteanu M, Ratziu V, Imbert- Bismut F, et al. The predictive value of Fibrotest vs. APRI for the diagnosis of fibrosis in chronic hepatitis C. Hepatology. 2004; 39:862–3.
28. Myers RP, Tainturier MH, Ratziu V, Piton A, Thibault V, Imbert- Bismut F, et al. Prediction of liver histological lesions with biochemical markers in patients with chronic hepatitis B. J Hepatol. 2003;39:222–30.
29. Thabut D, Trabut JB, Massard J, Rudler M, Muntenau M, Messous D, et al. Non-invasive diagnosis of large oesophageal varices with FibroTest in patients with cirrhosis: a preliminary retrospective study. Liver International. 2006:26:271–8.
30. Patel K, Gordon SC, Jacobson I, Hézode C, Oh E, Smith KM, et al. Evaluation of a panel of non-invasive serum markers to differentiate mild from moderate-to-advanced liver fibrosis in chronic hepatitis C patients. J Hepatol. 2004;41:935–42.
31. Talwalkar JA, Kurtz DM, Schoenleber SJ, West CP, Montori VM. Ultrasound-based transient elastography for the detection of hepatic fibrosis: systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2007;5:1214–20.
32. Ganne-Carrié N, Ziol M, de Ledinghen V, Douvin C, Marcellin P, Castera L, et al. Accuracy of Liver Stiffness Measurement for the Diagnosis of Cirrhosis in Patients with Chronic Liver Diseases. Hepatology. 2006;44:1511–17.
33. Yoneda M, Yoneda M, Fujita K, Inamori M, Tamano M, Hiriishi H, et al. Transient elastography in patients with non-alcoholic fatty liver disease (NAFLD). Gut 2007;56:1330–1.
34. Coco B, Oliveri F, Maina AM, Ciccorossi P, Sacco R, Colombatto P, et al. Transient elastography: a new surrogate marker of liver fibrosis influenced by major changes of transaminases. J Viral Hepat. 2007;14:360–9.
35. Sagir A, Erhardt A, Schmitt M, Häussinger D. Transient Elastography Is Unreliable for Detection of Cirrhosis in Patients with Acute Liver Damage. Hepatology. 2008;47:592–5.
36. Yeh WC, Li PC, Jeng YM, Hsu HC, Kuo PL, Li ML, et al. Elastic modulus measurements of human liver and correlation with pathology. Ultrasound Med Biol. 2002;28:467–74.
37. Vizzutti F, Arena U, Romanelli RG, Rega L, Foschi M, Colagrande S, et al. Liver stiffness measurement predicts severe portal hypertension in patients with HCV related cirrhosis. Hepatology. 2007:45:1290–7.
38. Kazemi F, Kettaneh A, N’kontchou G, Pinto E, Ganne-Carrie N, Trinchet JC, et al. Liver stiffness measurement selects patients with cirrhosis at risk of bearing large oesophageal varices. J Hepatol. 2006;45:230–5.
39. Foucher J, Chanteloup E, Vergniol J, Castera L, Le Bail B, Adhoute X, et al. Diagnosis of cirrhosis by transient elastography (FibroScan): a prospective study. Gut. 2006;55:403–8.
40. Lim AK, Taylor-Robinson SD, Patel N, Eckersley RJ, Goldin RD, Hamilton G, et al. Hepatic vein transit times using a microbubble agent can predict disease severity non-invasively in patients with hepatitis C. Gut. 2005;54:128–33.
41. Pandharipande PV, Krinsky GA, Rusinek H, Lee VS. Perfusion imaging of the liver: Current challenges and future goals. Radiology. 2005;234:661–73
42. Malik R, Afdhal N. Stiffness and Impedance: The New Liver Biomarkers. Clin Gastroenterol Hepatol. 2007;5:1144–6. Callewaert N, Van Vlierberghe H, Van Hecke A, Laroy W, Delanghe J, Contreras R. Noninvasive diagnosis of liver cirrhosis using DNA sequencer-based total serum protein glycomics. Nat Med 2004; 10: 429–34.
43. Callewaert N, Van Vlierberghe H, Van Hecke A, Laroy W, Delanghe J, Contreras R. Noninvasive diagnosis of liver cirrhosis using DNA sequencer-based total serum protein glycomics. Nat Med 2004; 10: 429–34.
44. Parsons CJ, Bradford BU, Pan CQ, Cheung E, Schauer M, Knorr A, et al. Anti-fibrotic effects of a tissue inhibitor of metalloproteinase-1 antibody on established liver fibrosis in rats. Hepatology. 2004;40:1106–15.
45. Murphy FR, Issa R, Zhou X, Ratnarajah S, Nagase H, Arthur MJ, et al. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: Implications for reversibility of liver fibrosis. J. Biol. Chem. 2002; 277:11069–76.
46. Iimuro Y, Nishio T, Morimoto T, Nitta T, Stefanovic B, Choi SK, et al. Delivery of matrix metalloproteinase-1 attenuates established liver fibrosis in the rat. Gastroenterology. 2003;124:445–58.
47. Siller-López F, Sandoval A, Salgado S, Salazar A, Bueno M, Garcia J, et al. Treatment with human metalloproteinase-8 gene delivery ameliorates experimental rat liver cirrhosis. Gastroenterology. 2004;126:1122–1133.
48. Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Nakatani T, et al. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology. 2001;34:745–50.
49. Terui Y, Saito T, Watanabe H, Togashi H, Kawata S, Kamada Y, et al. Effect of angiotensin receptor antagonist on liver fibrosis in early stages of chronic hepatitis C. Hepatology. 2002;36:1022.
50. Zhou X, Murphy FR, Gehdu N, Zhang J, Iredale JP, Benyon RC. Engagement of alphavbeta3 integrin regulates proliferation and apoptosis of hepatic stellate cells. J. Biol. Chem. 2004;279:23996–4006.
51. Yata Y, Gotwals P, Koteliansky V, Rockey DC. Dose dependent inhibition of hepatic fibrosis in mice by a TGF-beta soluble receptor: Implications for anti-fibrotic therapy. Hepatology 2002;35:1022–30.
52. Oakley F, Meso M, Iredale JP, Green K, Marek CJ, Zhou X, et al. Inhibition of inhibitor of kappaB kinases stimulates hepatic stellate cell apoptosis and accelerated recovery from rat liver fibrosis. Gastroenterology. 2005;128:108–120.
53. Hazra S, Xiong S, Wang J, Rippe RA, Krishna V, Chatterjee K et al. Peroxisome proliferator-activated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J Biol Chem. 2004;279:11392–401.
54. Wang BE. Treatment of chronic liver diseases with traditional Chinese medicine. J Gastroenterol Hepatol. 2000;15:E67–E70.
55. Sakaida I, Tsuchiya M, Kawaguchi K, Kimura T, Terai S, Okita K. Herbal medicine Inchin-ko-to (TJ-135) prevents liver fibrosis and enzyme-altered lesions in rat liver cirrhosis induced by a cholinedeficient L-amino acid-defined diet. J Hepatol 2003;38:762–9.
56. Shimizu I, Ma YR, Mizobuchi Y, Liu F, Miura T, Nakai Y, et al. Effects of Sho-saiko-to, a Japanese herbal medicine, on hepatic fibrosis in rats.Hepatology. 1999;29:149–60.
57. Zhang XL, Liu L, Jiang HQ. Salvia miltiorrhiza monomer IH764-3 induces hepatic stellate cell apoptosis via caspase-3 activation. World J Gastroenterol 2002;8:515–9.
58. Houglum K, Venkataramani A, Lyche K, Chojkier M. A pilot study of the effects of d-alpha-tocopherol on hepatic stellate cell activation in chronic hepatitis C. Gastroenterology. 1997;113:1069–73.
59. Mato JM, Camara J, Fernandez de Paz J, Caballeria L, Coll S, Caballero A, et al. S-adenosylmethionine in alcoholic liver cirrhosis: a randomized, placebo-controlled, double-blind, multicenter clinical trial. J Hepatol. 1999;30:1081–9.
60. Rambaldi A, Gluud C. Meta-analysis of propylthiouracil for alcoholic liver disease – a Cochrane Hepato-Biliary Group Review. Liver. 2001;21:398–404.
61. Rockey DC. Antifibrotics Therapy in Chronic Liver Disease. Clin Gastr and Hepatol. 2005;3:95–107.
62. Muddu AK, Guha IN, Elsharkawy AM, Mann DA.. Resolving fibrosis in the diseased liver: Translating the scientific promise to the clinic. Int J Biochem Cell Biol. 2007;39:695–714.
|
|
|
|
|
|